Confocal microscopy, most frequently Confocal Laser Scanning Microscopy (CLSM) or Laser Confocal Scanning Microscopy (LCSM), is an optical imaging technique for increasing the optical resolution and contrast of a micrograph by means of using a spatial pinhole to block out-of-focus light or glare in image formation.
- confocal microscopy
1. Confocal Microscopy
Confocal microscopy offers several advantages over conventional wide field optical microscopy, including the ability to control depth of field, elimination or reduction of background information away from the focal plane (that leads to image degradation), and the capability to collect serial optical sections from thick specimens whose thickness exceeds the immediate plane of focus (Figure 1).
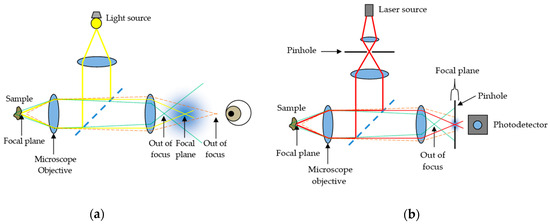
Figure 1. Comparison between (a) conventional optical microscopy and (b) confocal laser scanning microscopy.
In modern confocal systems, stacks of optical sections can be recorded over time, enabling the observation of time-dependent processes within 3D volumes (4D microscopy). In skin research, confocal techniques are commonly used for the in vivo investigation of surfaces, tissue structure, or cell cultures as well as for ex vivo histological studies. Among researchers, CLSM is becoming increasingly popular due to the relative ease with which extremely high-quality images can be obtained, where it is possible to observe the cellular size and shape, the epidermal structure, the dermal collagen morphology, and the keratin [1][8], serving as a tool for the analysis of equivocal skin lesions.
The spatial resolution of the system, which is the system’s ability to resolve two adjacent point objects in the lateral and axial directions, depends on the size of the illumination spot. The size of the illumination spot on the sample ranges from approximately 0.25 to 0.8 micrometers in diameter, which accounts for the lateral resolution (depending upon the objective numerical aperture), and 0.5 to 1.5 micrometers in depth at the brightest intensity, accounting for axial resolution [2][9]. The maximum depth within the sample from which optical sections with acceptable signal-to-noise ratio can be recorded depends on the imaging wavelength (excitation and emission), the working distance of the objective, and the optical density of the sample. In general, it is considered to be about 70 μm.
Regarding confocal microscope architectures, there are two outstanding families: slow scan, single beam confocal microscopes and faster Nipkow systems using a multiple beam approach. Single beam instruments scan the sample point by point in a raster pattern, working with galvanometer-driven mirrors. These instruments work with different laser lines for excitation: Argon ion lasers (488 and 514 nm), Argon–Krypton lasers (476, 488, 576, and 623 nm) and Helium-Neon lasers (543 and 632 nm). They use Photomultiplier Tubes (PMT) or hybrid detectors, ideally using one separate detection channel for each dye. On the other hand, Nipkow systems are equipped with a spinning disk behind the light source, containing an array of micro lenses or holes, which divide light into multiple beams simultaneously focused at the sample. These multiple beam instruments can work with normal arc-discharge lamps, and the signal can be captured in video rate with an ordinary charge-coupled device (CCD) camera. The former is better to image thick and dense tissue (no cross talk between multiple beams), and the latter is best for the investigation of rapid processes in living cells or tissue.
Looking at the number of different imaging modes in confocal microscopy, two basic operating modes can be differentiated: reflectance and fluorescence CLSM. They both have been used to conduct different research objectives in the field of dermatology.
2. Reflectance CLSM
Reflectance CLSM imaging uses a detection wavelength identical to the excitation wavelength. Here, light from reflection and scattering events is recorded, but no fluorescence signal at wavelengths longer than the excitation one is used. In this imaging mode, the image contrast derives mainly from refractive index changes, and the highest signal intensity usually originates from the transition between air and the sample surface. Thus, this mode is ideal for visualizing the topography of surfaces. Moreover, by using an immersion medium different from air (immersion oil, glycerin, or water), also structures lying under the sample surface can be observed. Since the reflectance configuration does not require staining of the sample, this mode can easily be used for in vivo imaging of skin surfaces and pathology, too.
3. Fluorescence CLSM
Fluorescence CLSM imaging uses a detection wavelength longer than the excitation one, since fluorescence emission displays a red shift characteristic for the respective fluorophore. This fluorescence is emitted either by skin autofluorophores, e.g., lipofuscin or NAD(P)H, or by fluorescent dyes that have been introduced into the sample following a specific staining protocol. The specificity of these staining techniques ranges from the general depiction of skin structure up to the highly specific staining of single target molecules in skin cells. Using a combination of fluorescent dyes with different emission characteristics, also different target structures can be simultaneously stained and visualized in parallel. In standard fluorescence microscopy, such investigations are mostly restricted to skin sections with a maximum thickness of ≈10 μm, since the superposition of stained structures in thicker samples impedes any discrimination of discrete labels.
The former modality, reflectance CLSM, which is applied in vivo, has become an important adjunct to clinical examination, dermoscopy, and histopathology assessment in the diagnosis and management of melanoma and other skin cancers [3][10]. For instance, it has been applied at the patient’s bedside for lateral margin detection because of its capability of exploring the skin at the cellular level, enabling the identification of tumor characteristics [4][11]. Additionally, the non-invasive nature of CLSM allows evaluations of skin lesions over time, thus enabling the investigation of dynamic physiological or pathological processes in vivo and at different time points [5][12]. Nevertheless, both modalities (especially, fluorescence CLSM) are applied ex vivo to tissue biopsies, and there is in-depth research on how to best preserve the properties of living tissue for as long as possible when preparing samples for visualization. For the 3D visualization of skin samples at high resolution, it is essential to preserve the skin structure in a near-to-life state. In the latter modality, fluorescence CLSM, the use of fluorescent dyes for skin staining even allows the 3D investigation of the structure in skin biopsies due to the possibility of optical sectioning in confocal systems. Hence, the CLSM investigation of samples stained with fluorescent labels enables the precise localization of cells or their constituents within an intact 3D environment [6][13]. In fact, ex vivo CLSM has also been used on freshly excised tumors in the operating room with different fluorophores allowing for tumor demarcation [4][11]. Some examples of contributions of CSLM imaging to the diagnosis of skin cancer are presented below, and in addition, summarized in Table 1.
Table 1. Contributions of Confocal Laser Scanning Microscopy (CLSM) to skin cancer diagnosis.
| Study | Year | Skin Cancer Type | No. of Lesions | Modality | Illumination Source |
No. of Images |
Axial Res. | Lateral Res. | SE/SP |
|---|---|---|---|---|---|---|---|---|---|
| Guitera et al. |
2009 | Melanoma, Nevus |
>300 | RCM, commercial CLSM |
Laser diode, 830 nm | >100 | 3–5 μm | 1 μm | Mel. Light- colored: 85%/84% Pigmented: 92%/65% |
| Guitera et al. |
2012 | Melanoma, BCC, Nevus, Pigmented facial macules, Other tumors |
>700 | RCM, commercial CLSM |
Laser diode, 830 nm | >100 | 3–5 μm | 1 μm | 100% 88.5% |
| Segura et al. |
2009 | Melanoma, BCC, SCC, Keratosis, Nevus | >150 | RCM, commercial CLSM |
Laser diode, 830 nm | >100 | 5 μm | 2 μm | 86.1% 95.3% |
| Ulrich et al. |
2008 | AK, perilesional healthy skin |
>20 | RCM, commercial CLSM |
Laser diode, 830 nm | 4–6 images |
3–5 μm | 1 μm | - |
| Horn et al. |
2008 | AK, perilesional healthy skin |
30 | RCM, commercial CLSM |
Laser diode, 830 nm | >50 | 3–5 μm | 1 μm | 93.34% 88.34% |
| Gareau et al. |
2008 | BCC | - | FCM, mosaicing |
Argon-ion laser, 488 nm |
36 × 36 images for a mosaic |
1.1 μm | 0.25 μm | - |
| Gareau et al. |
2009 | BCC, healthy skin | >40 | FCM, mosaicing |
Argon-ion laser, 488 nm |
45 confocal mosaics |
1.1 μm | 0.25 μm | 96.6% 89.2% |
| Abeytunge et al. | 2011 | BCC | 1 | FCM, strip mosaicing |
Argon-ion laser, 488 nm |
31 strips for a mosaic |
1.61 μm | 0.33 μm | - |
4. Contributions of CLSM Imaging
By means of reflectance CLSM, Pellacani et al. [7][14] have attempted to describe and characterize the cytological and architectural aspects of cell clusters in melanocytic lesions and to correlate them with routine histopathology.
In addition, Guitera et al. have conducted extensive research on the possibilities of reflectance CLSM evaluating melanomas [8][15] and basal cell carcinomas [9][16] by the implementation of a two-step scoring methodology. In the first study, they managed to improve the specificity compared with dermoscopy and, in addition, they were able to classify 100% of the non-biopsied control nevi population as benign. In the second study, they defined a multivariate algorithm to improve the diagnostic accuracy of basal cell carcinomas with very high sensitivity and specificity values. Under the microscope, they identified characteristic features of BCCs, such as telangiectasia and convoluted vessels, basaloid nodules, and epidermal shadowing corresponding to horizontal clefting.
Segura et al. [10][17] analyzed features of melanocytic and non-melanocytic skin tumors by reflectance CLSM and performed univariate and multivariate analyses to determine the association of Reflectance Confocal Microscopy (RCM) features with tumor types.
In addition, there are several studies on the assessment of Actinic Keratosis (AK), which is a precancerous condition that is very useful to detect in order to prevent skin cancer. Ulrich et al. [11][18] applied in vivo reflectance CLSM for the evaluation of AKs in correlation with routine histology. RCM features of AK included parakeratosis, architectural disarray, and keratinocyte pleomorphism. The presence of architectural disarray and cellular pleomorphism appeared to be the best predictor of AK. Horn et al. [12][19] also analyzed AKs under reflectance CLSM; the confocal images were rated by four independent derma oncologists, who were able to identify the distinct morphologic features from AKs. The overall sensitivity and specificity achieved were very high, showing the potential of reflectance CLSM as a non-invasive monitoring tool.
Ex vivo fluorescence CLSM is used more and more on a routine basis in the clinic. Freshly excised skin tumors are analyzed under fluorescence CLSM in the operating room. Different fluorophores, such as fluorescein, Nile blue, patent blue, methylene blue, and acridine orange, can be used at different wavelengths. However, acridine orange is the most commonly used because of its capability to provide an excellent contrast [4][11]. Most studies are performed on the commercial system VivaScope 2500® (MAVIG GmbH, Munich, Germany). The laser illumination wavelength is 488 nm. The depth is manually adjusted to image the surface. It provides optical sectioning of approximately 1.5 μm and resolution of approximately 0.4 μm. In addition, most studies have been performed in carcinomas, mostly in BCCs.
Gareau et al. have obtained confocal mosaics in excised skin lesions [13][20] and in particular of BCCs [14][21] to minimize the need for frozen histology. They used acetic acid as a reflectance contrast agent to enhance nuclear-to-dermis contrast. They compared the mosaics to histology, which requires 9 min instead of 20–45 min per excision for preparing frozen histology, and thus may provide a means for rapid pathology-at-the-bedside to expedite and guide surgery.
Abeytunge et al. [15][22] is another group of authors that have used fluorescence CLSM attempting to rapidly image larger areas of tissue. The limited field of view of high-resolution microscopes requires the merging of multiple images that are taken sequentially to cover a large area. This merging or mosaicing of images requires long acquisition and processing times, and it produces artifacts. They developed a methodology to reduce both time and artifacts, to image large areas of excised tissue with sub-cellular detail.