Emerging evidence indicates that the dysregulation of cellular redox homeostasis and chronic inflammatory processes are implicated in the pathogenesis of kidney and brain disorders. In this light, endogenous dipeptide carnosine (β-alanyl-L-histidine) and hydrogen sulfide (H2S) exert cytoprotective actions through the modulation of redox-dependent resilience pathways during oxidative stress and inflammation. Several recent studies have elucidated a functional crosstalk occurring between kidney and the brain. The pathophysiological link of this crosstalk is represented by oxidative stress and inflammatory processes which contribute to the high prevalence of neuropsychiatric disorders, cognitive impairment, and dementia during the natural history of chronic kidney disease.
- Hydrogen Sulfide,H2S,dysregulation of cellular redox homeostasis,chronic inflammatory processes
1. Introduction
Hydrogen sulfide (H2S) is a small gaseous molecule with profound biological effects within living organisms. It exerts key roles in cytoprotection, inflammation, vascular function, neurological systems, mitochondrial function, energy metabolism, and ageing [1][2][74,75]. However, H2S was originally known for its deleterious effects on health and the environment. First, in 1700, Italian physician Bernardino Ramazzini [3][76] described a severe ocular irritation and inflammation in sewer workers, caused by an unspecified volatile acid. Later, the chemical composition of H2S was elucidated and its association with ocular adverse effects and intoxication in sewer workers was recognized. For over a century, studies focused on its major toxic effects—e.g., inhibition of cytochrome c oxidase, carbonic anhydrase, monoamine oxidase, and sodium/potassium-ATPase (NaC/KC ATPase) [4][77]. The image of H2S was revolutionized when Kimura, in 1996, revealed its role as an endogenous neuromodulator [5][78]. Recently, H2S was classified as the third gasotransmitter along with NO and carbon monoxide (CO). H2S is enzymatically released in our body and it has a highly regulated metabolism. It is freely permeable to membranes and exerts specific physiological functions in several systems that can be mimicked by H2S donors applied exogenously [6][79]. In mammals, hydrogen sulfide (H2S) is primarily produced by two cytosolic pyridoxal-5′-phosphate-dependent enzymes, cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE), which use the substrates homocysteine and L-cysteine (Figure 1).
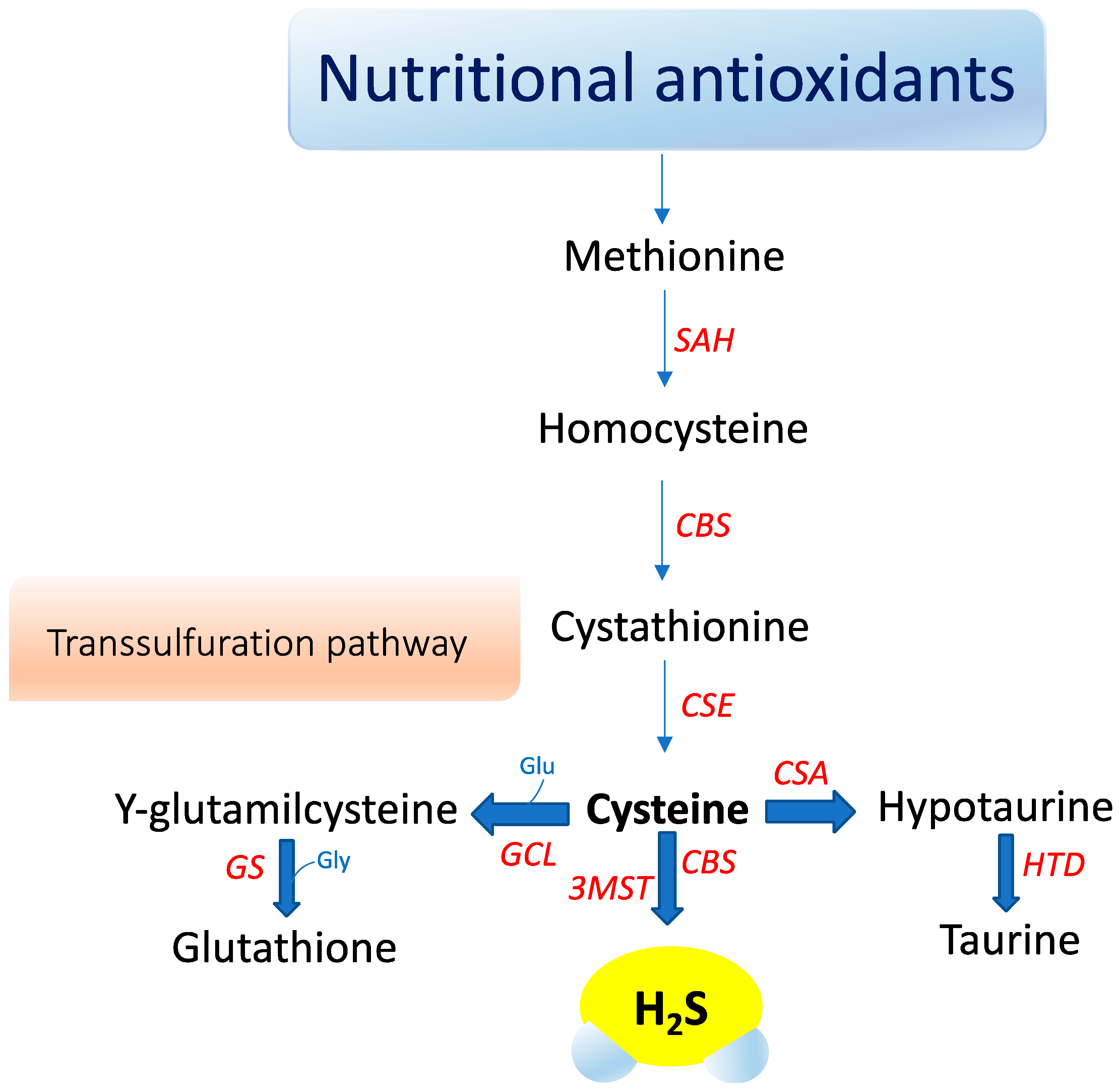
A third enzyme, 3-mercaptopyruvate sulfurtransferase (3-MST), catalyzes H2S production mainly in mitochondria by the conversion of 3-mercaptopyruvate to pyruvate and H2S [7][80]. Recently, an additional biosynthetic pathway has been described for the production of hydrogen sulfide from D-cysteine involving 3-mercaptopyruvate sulfurtransferase and D-amino acid oxidase that operates predominantly in the cerebellum and the kidney, i.e., within the KB axis [8][81]. D-cysteine is mainly adsorbed with food and derives from L-cysteine racemization during food processing. This novel pathway is of particular interest since supplementation with D-cysteine showed to protect renal cortex cells and cerebellum cells (i.e., KB axis) more efficiently than L-cysteine [8][81]. Furthermore, it was reported that gut microbiota would be another source of H2S that might influence health and function [8][81]. The contribution of each of these enzymes to net H2S production is dictated by its presence and relative tissue concentration, which varies in a cell-specific manner [8][9][10][81,82,83]. CBS is the major H2S-producing enzyme in the brain, while CSE is significantly expressed in the mammalian cardiovascular system and respiratory system and it seems to be the main H2S-forming enzyme in the liver, kidney, and pancreas [11][84].
Oxidative stress seems to influence CSE and CBS in a different manner. Reactive oxygen species appear to induce CSE expression, whereas they clearly suppress the transcription of the human CBS gene [12][85]. Eventually, H2S is metabolized to sulfite in the mitochondria by thiosulfate reductase, and then, is oxidized to thiosulfate and sulfate by sulfite oxidase. The sulfates are excreted in the urine [7][80]. Interestingly, urinary sulfates have been used as markers of H2S plasma levels and increased urinary sulfate concentrations have been correlated with a decreased risk of renal events in type 2 diabetic patients with nephropathy [13][86]. However, urinary sulfate and thiosulfate are not specific markers for endogenous H2S formation, and can also be the products of exogenous H2S production by sulfate-reducing bacteria in the gut [13][86]. H2S acts independently of any specific transporters by mechanisms not fully understood. S-sulfhydration, a novel posttranslational modification, is emerging as a mechanism responsible for many biological effects mediated by H2S [2][14][75,87]. H2S sulfhydrates protein thiol groups by transferring its sulfhydryl group to the cysteine residue of targeted proteins. Furthermore, H2S is oxidized in biological systems to polysulfides, which are now increasingly recognized as effectors of the H2S signaling mechanism [15][88]. H2S S-sulfhydration of enzymes, transcription factors, and ion channels has been described accounting for several protective effects of H2S, ranging from response to inflammation to cytoprotection [9][16][82,89]. For instance, H2S attenuates inflammation through the S-sulfhydration of Nuclear Factor-kappa B (NF-κB) [17][90]. H2S increases the antioxidative properties of cells by sulfhydration of Kelch-like ECH-associated protein 1 (Keap1), leading to its dissociation from Nrf2, which translocates in the nucleus and binds to the antioxidant response element (ARE) promoting antioxidant gene transcription, such as GCLM, GCLC, and glutathione reductase [18][91]. S-sulfhydration may be involved in the augment of the life span of Caenorhabditis elegans induced by H2S through sirtuins [19][92]. H2S may also inhibit mitochondria ROS production through sulfhydration of p66Shc [18][91]. In this regard, gut microbiota has attracted considerable interest for its role in microbial-mediated ROS generation, which might influence many signaling and homeostatic processes. Notably, lactobacilli ROS-mediated signaling has been described to induce Nrf2, opening the prospect that probiotic bacteria may elicit beneficial effects on disease states that involve Nrf2, including diabetes and neurodegenerative diseases [20][93]. In particular, in the (nod-like receptor) NLR family, the nod-like receptor pyrin domain-containing 3 (NLRP3) inflammasome has been reported to play a pathogenic role in the initiation and progression of metabolic and neurodegenerative diseases [21][22][94,95]. Recently, in vitro and in vivo studies showed that H2S mitigated lipopolysaccharide (LPS)-induced sepsis against oxidative stress and inflammation damage mediated by the NADPH oxidase 4 (Nox4) pathway [23][96], inhibiting the vicious cycle of NLRP3 inflammasome and oxidative stress in human retinal pigment epithelial cells [24][97] and in hypertensive rats [25][98]. In addition, H2S mediated effects on neuroinflammation and Aβ1-42 production by suppressing the activation of STAT3 and cathepsin S [26][99]. H2S suppresses oxidative stress-induced mtROS production and NLRP3 inflammasome activation via S-sulfhydrating c-Jun at cysteine 269 in macrophages [27][100].
2. H2S Redox Signaling and Resilience
Emerging evidence has highlighted the crucial role of H2S in maintaining redox homeostasis, which occurs by modulating levels of cellular antioxidant enzymes and increasing the expression of the transcription factor nuclear erythroid-related factor 2 (Nrf2) during oxidative stress. The latter is an intracellular excess of reactive oxygen species (ROS) relative to depletion of antioxidant capacity of the cell [28][230]. Interestingly, Nrf2 is a master regulator of redox cellular stress response in various pathological states [29][30][1,231]. Under physiological conditions, Nrf2 is localized in the cytosol and regulated by its inhibitor Kelch-like ECH-associated protein 1 (Keap1). Recently, much evidence has demonstrated that H2S induces cytoprotection against oxidative stress by the stimulation of Nrf2, which accumulates and translocates into the nucleus where it binds to the antioxidant response element (ARE) inducing the transcription of multiple target genes, including phase II detoxification enzymes such as NAD(P)H: quinone oxidoreductase 1 (NQO1), heme oxygenase 1 (HO-1), thioredoxin, γ-glutamylcysteine synthetase, and glutathione S-transferase (GST) [31][232]. In line with this observation, Nrf2 encodes the vitagene antioxidant pathway which exists to counteract different forms of stress (e.g., oxidative, environmental, and mitochondrial stress). Vitagenes include heat shock protein 70 (Hsp70), heme oxygenase 1 (HO-1), γ-glutamylcysteine synthetase (γ-GCs), thioredoxin (Trx), and sirtuins (SIRTs) (Figure 3) [29][32][33][34][1,233,234,235] as biomarkers for stress adaptation, cross-tolerance, and resilience underlying hormesis or preconditioning [35][236] (Figure 3).
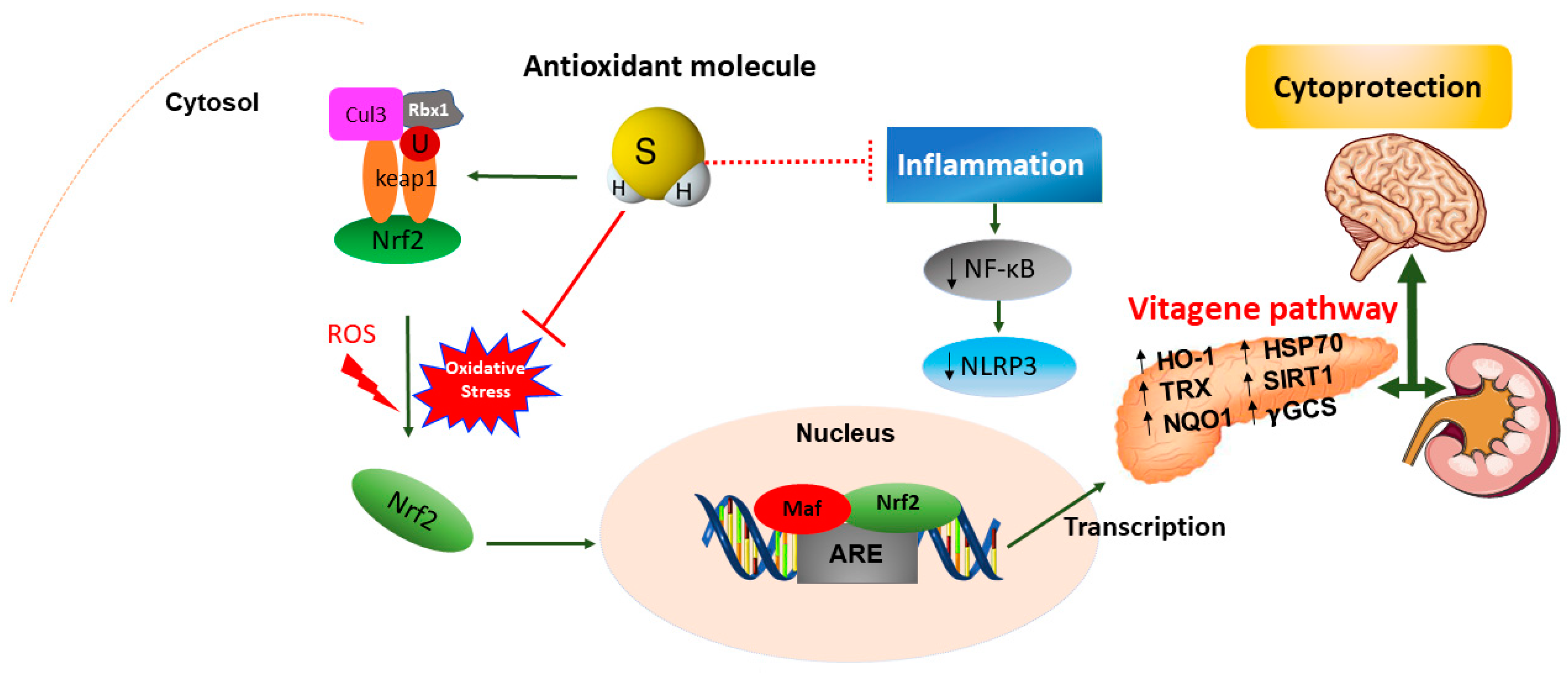
In addition, recent in vivo studies reveal the nephroprotective effects of H2S on renal tissue through upregulation of antioxidant proteins and anti-inflammatory cytokines, as well as the expression of eNOS and iNOS via induction of the Nrf2/HO-1 pathway in renal injury [36][302] and in the spinal cord of rats [37][303]. Moreover, H2S could attenuate high glucose-induced myocardial injury in rat cardiomyocytes by suppressing the Wnt/β-catenin pathway and upregulating the expression of HO-1 and NQO1 [38][304]. In addition, H2S protected renal tissue against ischemia-reperfusion injury-induced lipid peroxidation, inflammation, and apoptosis, which may be attributed to the upregulation of HSP 70, HO-1, and HSP 27 [39][305]. H2S reduces myocardial fibrosis in diabetic rats, which is related to the inhibition of protein kinase Cα (PKCα), upregulation of HSP70 expression [40][306], and downregulation of the JAK/STAT signaling pathway [41][307]. Finally, H2S protects cells from oxidative stress [42][308] and delays programmed cell death by increasing the levels of antioxidant glutathione and HO1 expression [43][309]. Taken together, the data above convincingly indicate the crucial role of H2S as a potent antioxidant molecule that, at low concentrations, induces protective actions exploited through the redox modulation of the Nrf2 vitagene signaling pathway which may provide a novel potential therapeutic approach to confer resilience against oxidative stress, inflammation as well as apoptosis during pathological conditions such as diabetes and related complications.