Neurodegenerative pathologies are commonly characterized by the misfolding, oligomerization and accumulation of toxic species such as Aβ in AD, α-Syn in PD, and the prion protein in CJD. In addition, it appears that a major source of pro-inflammatory diffusible signals associated with brain neuroinflammation originates from peripheral organs and systems such as the gastrointestinal (GI) tract microbiome. Bacterial components such as LPS, which can enter the bloodstream, stimulate systemic pro-inflammatory responses in the host including the CNS. At the cellular and molecular levels, LPS is able to induce the release of inflammatory mediators and eventually induce synaptic loss, which can lead to cognitive impairment via microglial activation, generation of reactive oxygen species (ROS) and oxidative stres. Finally, it appears that signals released by bacteria can modulate amyloid formation and activate pro-inflammatory responses in the brain, suggesting a strong interplay between the microbiome and neuroinflammation in neurodegenerative diseases initiation and progression.
- Amyloid, Alzheimer Disease, microbiota,
Please note: Below is an entry draft based on your previous paper, which is wrirren tightly around the entry title. Since it may not be very comprehensive, we kindly invite you to modify it (both title and content can be replaced) according to your extensive expertise. We believe this entry would be beneficial to generate more views for your work. In addition, no worry about the entry format, we will correct it and add references after the entry is online (you can also send a word file to us, and we will help you with submitting).
Definition
Neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and Creutzfeldt–Jakob disease (CJD) are brain conditions affecting millions of people worldwide. These diseases are associated with the presence of amyloid-β (Aβ), alpha synuclein (α-Syn) and prion protein (PrP) depositions in the brain, respectively, which lead to synaptic disconnection and subsequent progressive neuronal death. A body of research suggests a potential association between host microbiota, neuroinflammation and dementia, either directly due to bacterial brain invasion because of barrier leakage and production of toxins and inflammation, or indirectly by modulating the immune response.
1. The Burden of Neurodegenerative Diseases
Currently, nearly 50 million people worldwide suffer from neurodegenerative diseases (NDDs), mainly dementia, and this number is expected to reach 152 million by 2050 [1]. It is noteworthy that we are experiencing a shift in global demographics towards a large elderly population, which is increasing the prevalence of neurodegeneration worldwide and the financial burden associated with these diseases (e.g., medication, nursing care). For example, it is estimated that in the USA alone more than 5 million people aged 65 or older suffer from AD, and the costs of treating the disease are estimated at over US$180 billion per year [2,3][2][3].
In recent years, considerable progress has been made regarding the pathogenesis, diagnosis and treatment of Alzheimer’'s disease (AD), Parkinson’'s disease (PD) and Creutzfeldt-Jakob disease (CJD). However, these pathologies remain debilitating and fatal conditions, with significant negative medical, economic and social impacts. To date, there are no effective therapeutic approaches to prevent, delay or reverse these disorders, which start with cognitive loss and alterations of neurovegetative functions and progress towards language deficit, memory loss, motor difficulties and ultimately death [4]. These neurodegenerative diseases are associated with neuronal loss in several regions of the brain, such as the frontal cortex, hippocampus and basal ganglia. AD and PD can be classified as either “"early-onset, genetic”" (also known as “"familial”) or “") or "late-onset, sporadic”" [5]. Most significantly, the late-onset forms are more prevalent and are considered to be the main cause of dementia and motor disease in the elderly population [6].
The most common neurodegenerative disease is AD, which is mainly characterized by marked cognitive dysfunction, impairment in the formation of new memories, and synaptic failure [7,8][7][8]. AD hallmarks include intracellular neurofibrillary tangles and the extracellular deposition of senile plaques that are mainly constituted by amyloid-β (Aβ) peptide [9]. Aβ is able to rupture the neuronal plasma membrane by the formation of pores leading to cytoplasmic leakage and cell death [10,11][10][11], by either direct lipid disruption or by its interaction with ion channels in the membrane [12,13][12][13]. Current research suggests that late-onset AD is mostly determined by environmental factors such as toxins, trauma and diet [14]. However, the underlying mechanism of action has not been completely elucidated yet.
The second most common neurodegenerative pathology is PD. In 2016, 6.1 million people worldwide were living with a diagnosis of PD, and it was estimated that 10 million people would be suffering from this disease by 2020 [15]. The prevalence of PD ranges from 100 to 200 cases per 100,000 people [16], and it affects nearly 3% of the population older than 65 years of age [17]. PD is mainly characterized by motor symptoms including bradykinesia, rigidity, tremor, postural instability, dysphagia and axial deformities, and non-motor symptoms such as cognitive dysfunction, sleep disorder, depression, anxiety, apathy, pain and dementia [17,18][17][18]. PD has been well described as the intraneuronal deposition of alpha synuclein (α-Syn), which contributes to the generation of protein inclusions known as Lewy bodies [19]. It is widely known that the loss of dopaminergic neurons in the substantia nigra pars compacta is the landmark physiopathological sign of the disease [18]. Due to the deleterious consequences of α-Syn, it has been considered a strategic target for future therapies to ameliorate the symptoms and slow down the progression of the disease.
Another relevant group of neurodegenerative disorders are prion diseases. There are three types of human prion disorders: sporadic, genetic and acquired [20]. The most common form of prion disease is sporadic CJD, a fatal pathology caused by misfolded prion proteins [20]. CJD is responsible for 85% of diagnosed prion disease cases, with a reported incidence of 1–2 cases per million people per year worldwide, and about 350 new annual cases in the United States [21]. The onset of CJD occurs in patients older than 67 years of age [20]. The main features of CJD and prion diseases are spongiform changes in gray matter, gliosis and neuronal death [22,23][22][23]. The most reported symptoms for CJD are progressive dementia, behavioral and cognitive impairment, insomnia, movement disorder and ataxia [24].
As a common feature, all neurodegenerative diseases seem to be associated with protein misfolding that leads to synaptic alterations, neuronal membrane damage and neuroinflammation. In addition, it has been recently suggested that microbial components, such as the ones present in the host microbiome, may also be actively involved in modulating neuroinflammation and protein misfolding. Therefore, in the present review we focus on the emerging hypothesis regarding the role of the host microbiome and its dysregulation in the onset of neurodegeneration, via (i) the entry of microbial cells, toxins and outer membrane vesicles directly into the brain, and (ii) the induction and maintenance of a systemic chronic inflammatory state. Furthermore, we discuss the involvement of associated systems such as the oral microbiome and bile, and potential routes of entry for bacteria and toxins into the central nervous system (CNS).
2. Protein Misfolding and Its Accumulation in Neurodegenerative Diseases
Neurodegenerative pathologies are commonly characterized by the misfolding, oligomerization and accumulation of toxic species such as Aβ in AD, α-Syn in PD, and the prion protein in CJD [24,25][24][25]. These protein alterations trigger neuronal degeneration and dysfunction and drive the progression of each particular disease [25]. For instance, there is abundant evidence demonstrating that Aβ peptide accumulation initiates and promotes AD. Aβ is mainly detected in the extracellular matrix in the brain and cerebrospinal fluid (CSF) at nanomolar concentrations, and is widely accepted as the main neurotoxic agent in the disease [26]. It is believed that early manifestations of AD are associated with the synaptotoxic effects produced by soluble oligomeric forms of Aβ [27]. The existence of mutations in genes for the amyloid-β precursor protein (AβPP) (chromosome 21) and presenilin 1 (chromosome 14) and 2 (chromosome 1) have also been reported in some AD patients, providing further evidence that Aβ is an important factor in the development of AD [28].
A well-accepted hypothesis for AD generation is that monomers of Aβ oligomerize, first forming low molecular weight species referred to as oligomers [27], which have been found to be highly neurotoxic to the membrane [13]. There is no clear consensus about the most toxic species, but there is an agreement that starting from dimers up to 56 kDa, oligomers are the most important causal agents in the disease [29,30][29][30]. These peptides/proteins can associate and damage the cell membrane, affecting neuronal function. The semipermeable property of the membrane is critical for cellular homeostasis, and the resulting Aβ-induced leakage of cellular components, as well as the non-regulated calcium influx into the cell, will turn into synaptotoxicity [13,31][13][31]. All the available evidence points to the idea that Aβ toxic events are multiple and that one/several of them might serve as a therapeutic target. Likewise, PD is mainly characterized by the formation of intracellular Lewy bodies in dopaminergic neurons. These structures are mostly formed by intracellular accumulation of α-Syn [24], a 140-residue protein encoded by the Synuclein Alpha (SNCA) gene that drives neurodegeneration. Prion diseases, on the other hand, have spongiform vacuolation, gliosis, neuronal loss and deposition of amyloid molecules immune-positive for prion protein (PrP) as hallmarks of the disease [24]. Thus, prion disorders are caused by the misfolded form of the prion protein, denoted prion protein scrapie (PrPSc) [32]. The toxic misfolded PrPSc has a high content of β-sheet in its secondary structure, which generates a highly hydrophobic and insoluble protein with a high tendency to aggregate and form amyloid structures [24,32][24][32].
3. Protein-Induced Membrane Damage as a Central and Ubiquitous Player in Neurotoxicity
As discussed above, it is widely accepted that the accumulation of misfolded proteins is an important hallmark for AD, PD and CJD. Most importantly, these proteins are capable of inducing membrane damage in the brain by assembling monomers into non-selective ion pores and subsequently inserting them into a variety of cell membranes. For example, α-Syn oligomers increase the permeability of cell membranes in distinct types of neurons [33]. Additionally, α-Syn is also known to form pores in phospholipid bilayers found in mitochondria, inducing a complex series of multilevel conductance reminiscent of the effects of Aβ in hippocampal membranes [34]. α-Syn insertion into bilayers is facilitated by cardiolipin, an important phospholipid present in mitochondrial membranes [34]. As mitochondria are key organelles for cell energetic and ionic homeostasis, membrane alterations by oligomeric proteins can result in important alterations of cell viability.
Recent data using nanoelectrospray and mass spectrometry have shown that Aβ42 oligomerizes and forms β-barrel structure hexamers, which can be stabilized by the addition of lipids [35]. A similar situation is observed for toxic oligomers of PrP, associated with cellular membranes, where they might induce fast and prolonged toxic effects [36]. Studies in lipid bilayers, for example, have indicated that PrP oligomers cause a rapid and large increase in the permeability of the membrane, whereas monomeric forms cause no detectable leakage [36]. More recent studies using calcein-leakage assays showed that soluble prion oligomers are capable of producing leakage in negatively charged vesicles [37]. Studies at the nanometer level with atomic force microscopy showed that a fragment of the human PrP spanning residues 106–126 (PrP106–126) disrupted the intrachain conformation of phosphatidylcholine lipids [38]. All these results support the idea that, similar to Aβ and α-Syn, PrP oligomers can disrupt cell membranes. Further data regarding the relevance of these molecules in disease pathogenesis were obtained using the PrP27–30 fragment extracted from the brains of terminally ill golden Syrian hamsters infected with the 263K scrapie strain [39]. Interestingly, the electrophysiological recordings carried out with PrP resembled membrane responses obtained with Aβ in native neurons, including high variability on the amplitude of the unitary response and some spontaneous membrane breakages [11]. The responses showed a multistate conductance current, with at least one amplitude near 80 pS, a reversal around 0 mV and dependency on cation concentration (Na+ and K+). In addition, using the recombinant fragment of PrP (PrP90–231) a similar dependence on calcium was shown. In sum, AD, PD and prion diseases are associated with membrane alterations, increases in calcium permeability and ionic dyshomeostasis, which contribute to neurodegeneration. Most importantly, potentiation of local brain factors with other peripheral inflammatory mediators (such as those derived from a dysbiotic gut) may be associated with the progression of neurodegenerative diseases.
4. Neuroinflammation as a Common Factor across Neurodegenerative Diseases
As previously noted, synaptic and cellular alterations mediated by misfolded protein accumulation are the main hallmarks across NDDs [40]. However, all these diseases also share the common ground of displaying an increased inflammatory response in the brain, known as neuroinflammation. This process involves the activation of resident microglia and astrocytes that produce cytokines, chemokines and other inflammatory molecules within the CNS. Many of these markers are universal across NDDs, supporting the idea of a common neuroinflammatory profile across these diseases. Some of these common neuroinflammation mediators are chitotriosidase 1 (CHIT1), chitinase-3-like protein 1 (YKL-40), the glial fibrillary acidic protein (GFAP) and important pro-inflammatory cytokines, such as interleukin-1β (IL-1), IL-6 and tumor necrosis factor α (TNF-α) [41,42][41][42].
In general, for proteinopathies such as AD, PD and prion diseases, it has been shown that neuroinflammation can be directly induced by amyloids. In the context of AD, not only do reactive microglia colocalize with amyloid deposits in situ, but also in vitro Aβ oligomers have been shown to directly induce microglial activation [43,44,45,46,47][43][44][45][46][47]. Characterization of inflammatory molecules in CSF and plasma from AD patients has shown increased levels of pro-inflammatory cytokines such as IL-1β, IL-6 and TNF-α [42] and increases in the macrophage colony-stimulating factor, which has been described as a microglial activator [48]. Similarly, animal models of AD such as TgAPPsw and PSAPP transgenic mice also show an increase in a pro-inflammatory profile characterized by cytokines IL-1, IL-6 and TNF-α, and the granulocyte macrophage colony stimulating factor. This observation is consistent with in vitro studies using microglial cell cultures exposed to Aβ42 [48]. IL-12 and IL-23 were produced by microglia in AD transgenic mice models (APP/PS1), and the genetic ablation of these cytokines resulted in a decrease in cerebral amyloidosis [49].
In PD, microglia activation in the SNpc and striatum is well documented in murine models [50]. However, most of the microglial activation by α-Syn misfolding has been attributed to a deleterious pro-inflammatory response that is related to dopaminergic neuron degeneration [50,51][50][51]. As for AD, the cytokine profile in PD brains is characterized by the release of pro-inflammatory molecules IL-1β, IL-6, IL-12, interferon gamma (IFN-γ) and TNF-α [50,52][50][52]. Therefore, microglial response is an early marker of neuroinflammation in NDDs and seems to be the first mediator in the innate immune reaction in the CNS in these pathologies.
Studies in human prion diseases indicate that microglial activation correlates with the onset of the clinical signs and that its magnitude depends on prion strain [53,54][53][54]. Nevertheless, clustering analysis of neuroinflammatory gene expression performed in different brain regions of prion-infected mice suggested that astrocyte function is altered before microglia activation [55]. In this sense, transient prion neuroinflammation events show only partial similarity with the microglia degenerative phenotype reported in animal models of other NDDs, where microglial activation precedes astrogliosis. Regarding cytokine profiles in prion-induced neuroinflammation, similar markers to AD and PD such as TNF-α, IL-1β and particularly IL-1α are significantly increased in brain tissue from infected mice and CJD patients [56,57][56][57].
Since microglia are a key element in neuroinflammatory responses, and a predominantly inflammation-linked cytokine profile is found in AD, PD and prion diseases, microglial activation in these pathologies is considered to be associated with the pro-inflammatory M1 phenotype [58]. Nevertheless, anti-inflammatory cytokines such as IL-4, IL-10 and IL-13 are increased and have been detected in the striatum of PD patients [50]. Furthermore, increased levels of IL-4 and IL-10 have been found in CSF samples from AD [59] and CJD patients [60,61][60][61]. Due to recently developed one-cell transcriptome analyses, it has been possible to separately define specific phenotypic changes in microglia, astrocytes and neurons. In AD, these analyses have revealed microglial subpopulations with a distinctive molecular signature different from the classical M1 and M2 phenotype, which has led to the concept of disease-associated microglia (DAM) [62,63][62][63]. Two main receptors have been identified as key regulators in the generation of these particular phenotypes in neurodegenerative diseases: Toll-like receptors (TLRs) and triggering receptors expressed on myeloid cells-2 (TREM2) [62]. TREM2 interacts with two adaptor proteins, DAP12 and DAP10 [62]. Mutations in these proteins have been linked to AD, PD and other misfolding-related neurodegenerative disorders [64]. Even though the role of TREM2 signaling in neurodegeneration has not been defined, since both protective and harmful responses have been described, TREM2 has a clear role in the induction of the DAM phenotype [62]. For instance, in 5xFAD mice (a transgenic model of AD), single-cell transcriptome analyses revealed the existence of two DAM microglia clusters. Both clusters exhibited downregulation of homeostatic genes and upregulation of a particular signature that includes TREM2. In addition, TREM2 can act as a receptor for Aβ [65,66][65][66]. Similar microglial disease-specific phenotypes, distinguishable from the classic M1 phenotype induced by lipopolysaccharide (LPS), have been observed in other neurodegenerative disorders such as amyotrophic lateral sclerosis and multiple sclerosis [55,62][55][62]. Nevertheless, it is important to highlight that probably both elements, classical M1 and DAM, might be relevant in the progression of these diseases, with M1 contributing to the detrimental neuroinflammatory effects [62].
On the other hand, TLRs include 13 members that recognize different molecular patterns associated with pathogens, with LPS being one of the classical TLR inductors [62]. Besides pathogens, misfolded proteins may induce TLRs. In this sense, both α-Syn and Aβ have been described as TLR ligands [67,68][67][68]. Furthermore, some bacterial metabolites have also been described as ligands for TLR2 and TLR4 [69]. TREM2 has been also found to bind LPS, which is the most well-characterized bacterial-derived molecule in neurodegenerative disease models [70]. LPS is able to activate pro-inflammatory responses and contribute to detrimental effects in AD, PD and Huntington’'s disease [71]. In the early stages of prion disease in ME7 prion strain-infected mice, LPS injection leads to exacerbated impairment in locomotor and cognitive functions [72]. Overall, LPS inoculation experiments suggest that bacteria-derived products could accelerate disease progression and contribute to neuronal decline.
Overall, activation of microglia is linked to the production of pro-inflammatory cytokines known to have deleterious effects when increased in tissues, including the brain [73]. IL-1β and TNF-α are able to reduce synaptic plasticity after acute application in brain slices [73]. Additionally, neurons express cytokine receptors that stimulate the mitogen-activated protein kinase (MAPK) family and lead to a reduction in synaptic efficiency [73]. Several calcium signaling mechanisms, including N-methyl-D-aspartate receptors (NMDARs), inositol trisphosphate receptor, ryanodine receptors and voltage-sensitive Ca2+ channels (VSCCs), may be modulated by cytokines in neurons. In this sense, increased levels of TNF-α can trigger calcium release from intracellular compartments and increase the expression of L-type VSCC. Neurons also express IL-1RAcPb, a neuron-specific IL-1 receptor accessory protein relevant for IL-1β binding that has been linked to an alternative phosphorylation pathway through Src phosphorylation, which is able to enhance Ca2+ influx through NMDAR activation [73].
In conclusion, AD, PD and prion diseases show early features of neuroinflammation that can be directly linked to misfolded protein deposition, which in turn triggers a specific microglia- and astrocyte-activated phenotype. However, external sources of neuroinflammation, different from those directly related to misfolded proteins, are also able to increase neuronal damage. In this sense, systemic inflammation could play an important role in the onset and maintenance of neuroinflammation; thus, the most recent evidence regarding the association between oral and gut microbiota and the promotion of an inflammatory state will be discussed, as well as its potential link with neurodegenerative diseases.
5. Human Microbiome Dysbiosis as a Source of a Systemic Chronic Inflammatory State
It is currently known that humans are inhabited by a wide and diverse range of microorganisms including bacteria, viruses and fungi, among others. These microorganisms, conjunctively known as the human microbiome, are compartmentalized in different areas of the human body such as the oral cavity, skin and gut; thus, each one of these “"niches”" holds a specific microbial composition. It is currently believed that we carry around more microbial cells on a daily basis than our own human cells [74]. Recently, it has been demonstrated that an overall healthy microbiome is crucial for maintaining homeostasis, and that imbalances in microbiota composition (i.e., dysbiosis) can lead to disease in many tissues and organs [75]. Systemic diseases such as cardiovascular disease, diabetes mellitus, rheumatoid arthritis and obesity are all believed to have a direct association with microbiome dysregulation, either via the direct effect of certain pathologic species or due to modulation of the host inflammatory response [76].
6. Direct and Indirect Effects of Microbiota on the Brain: Role of Barrier Evasion and Permeability
The CNS is one of the tissues that benefits from a degree of antigen tolerance, also known as immune privilege. This characteristic is mainly due to the presence of the blood–brain barrier (BBB), which separates the CNS from the systemic immune response and protects the brain and spinal cord from acute inflammatory mediators, which could induce more damage than immune control. This control is not only exerted by the BBB but also by the blood-cerebrospinal fluid barrier (BCSB) and the arachnoid barrier [40]. These barriers also explain why antigen emergence within the brain or spinal cord does not generate a peripheral immune response. The BBB is a complex and highly regulated exchange interface, composed of pericytes, astrocytic processes and nearby neurons adjacent to capillaries. It works as a carrier, an enzymatic barrier, a paracellular barrier (due to endothelial junctions) and a cerebral endothelium [209,210][77][78]. During systemic inflammation, both disruptive and non-disruptive changes in the BBB can be observed. Although no visible changes are produced with non-disruptive BBB damage, the changes in BBB physiology might alter astrocyte function and cytokine production, and higher levels of pathogen invasion can be produced [210][78]. Moreover, under non-disruptive alterations, very few molecules can cross the barrier. On the other hand, during disruptive events such as those induced by bacteria-derived LPS, histological and anatomical changes can be observed with strong alterations in permeability. In several neurodegenerative diseases such as AD, the BBB is also affected and its role in CNS permeability is compromised. In the case of AD, abnormal clearance of Aβ and an increased BBB permeability allowing the entrance of pro-inflammatory molecules into the brain are observed [210][78]. In a global systematic context, three membranous locations are critically important because of their physicochemical properties: membranes of the gastrointestinal–blood barrier, of the BBB, and finally the semipermeable neuronal membrane. It has been proposed that because of an increased leakage in both the gastrointestinal–blood barrier and the BBB in AD (and perhaps other NDDs), these pathologies might be considered as “"defective barrier”" diseases [69,211][69][79]. This increased permeability would facilitate the entry of bacterial cells, bacterial molecules and peripheral inflammatory mediators into the brain that subsequently would exacerbate local neuroinflammation via the mechanisms mentioned previously.
7. Microbiome Dysbiosis and Neuroinflammation: A Complex “"Toxic”" Mixture Affecting the Brain during NDD
As discussed throughout this review, it appears that a major source of pro-inflammatory diffusible signals associated with brain neuroinflammation originates from peripheral organs and systems such as the gastrointestinal (GI) tract microbiome. Bacterial components such as LPS, which can enter the bloodstream, stimulate systemic pro-inflammatory responses in the host including the CNS. At the cellular and molecular levels, LPS is able to induce the release of inflammatory mediators and eventually induce synaptic loss, which can lead to cognitive impairment via microglial activation, generation of reactive oxygen species (ROS) and oxidative stress [69] (Figure 1). It has been proposed that bacterial LPS may be involved in neuroinflammation associated with amyloid fibril formation in AD [212][80], suggesting that LPS acts as a promoter of Aβ fibrillogenesis in a time-dependent manner, possibly through a heterogeneous nucleation mechanism. It has also been shown that a single intraperitoneal injection of LPS increases Aβ42 levels and astrocyte activation in critical brain regions such as the cerebral cortex and hippocampus [101][81]. In addition, LPS affected memory in mice, suggesting the development of brain dysfunction [101][81]. These negative actions of peripheral LPS on amyloidogenesis, memory function and neuronal death were inhibited by sulindac, an anti-inflammatory agent, supporting the role of peripheral inflammation in AD pathology. Interestingly, it was shown that inflammatory cytokines such as IL-1β and TNF-α can increase the expression of APP and the formation of Aβ [213,214][82][83].
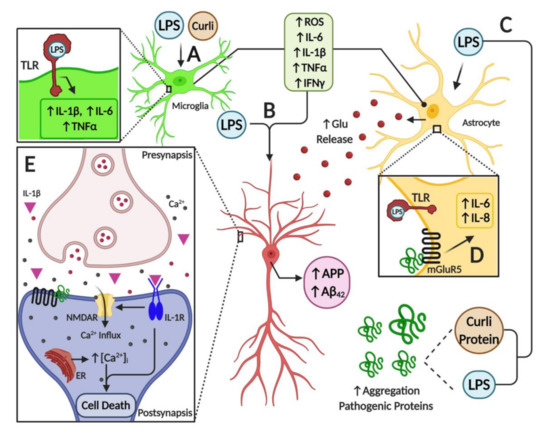
Illustration of neuroinflammatory mechanisms mediated by microbiome-derived products in nervous tissue. (
) Toll-like receptors (TLRs) expressed in glial cells are activated by LPS, triggering the activation of astrocytes and microglial cells. This activation induces an inflammatory response by overexpression and release of pro-inflammatory cytokines such as IL-6, IL-1β, TNF-α and IFN-γ, and by an increase in oxidative stress due to the generation of reactive oxygen species. Furthermore, bacterial amyloid proteins (curli) activate glial cells and induce the expression of pro-inflammatory mediators. (
) Pro-inflammatory mediators, together with LPS, increase the expression of the amyloid precursor protein (APP), and the deposition and misfolding of Aβ peptide. (
) Both LPS and curli are able to increase the deposition and aggregation of pathogenic proteins. (
) In astrocytes, among other cell types, activation of mGlurR5 receptor by pathogenic proteins triggers the overexpression of pro-inflammatory cytokines such as IL-6 and IL-8, which worsen the inflammatory milieu in the brain. Moreover, a high level of pro-inflammatory mediators leads to increased levels of the neurotransmitter glutamate, furthering ionic dyshomeostasis and augmenting neuronal excitotoxicity. (
) Finally, mGluR5 activation by pathogenic proteins induces the release of calcium from the endoplasmic reticulum, leading to ionic and mitochondrial dyshomeostasis, which results in neuron death. Furthermore, the activation of IL-1R in neurons by the binding of IL-1β cytokine amplifies the activity of NMDARs and mediates the inflammatory response via p38 MAPK. Overall, these alterations stimulate endoplasmic reticulum (ER) Ca
release through ryanodine receptors and IP3 receptors, which trigger ER stress and mitochondrial fragmentation leading to synaptic failure and neuronal apoptosis.
Furthermore, several studies have shown the interplay between toxins released by bacteria and neurodegeneration. For example, some Enterobacteria species may release amyloid peptides that alter the aggregation of α-Syn in the brain [215][84]. Another study also showed that when aged rats were exposed to curli-producing E. coli, an increased neuronal α-Syn deposition in both the gut and brain was observed; furthermore, animals also showed enhanced microgliosis and astrogliosis compared to those exposed to control bacteria unable to synthesize curli [216][85]. Rats exposed to curli also showed a higher expression of TLR2, IL-6 and TNF-α in the brain [216][85]. Overall, it appears that signals released by bacteria can modulate amyloid formation and activate pro-inflammatory responses in the brain, suggesting a strong interplay between the microbiome and neuroinflammation in neurodegenerative diseases (Figure 1).
The potential link between bacterial-derived products and neurodegeneration is strengthened by several other studies. For example, a reduction of several Aβ species in the brain and blood was detected in APPPS1 transgenic mice in the absence of gut microbiota [149][86]. Therefore, the presence of a GI germ-free condition reduced cerebral Aβ amyloid pathology in diseased mice when compared to control mice with control intestinal microbiota. Furthermore, the colonization of germ-free APP transgenic mice for 8 weeks with microbiota from conventionally raised APP transgenic mice increased Aβ42 levels [149][86]. Overall, these results support the idea that the GI microbiota is involved in the development of Aβ pathology in the brain, as well as the existence of pro-inflammatory mediators with the ability to enter the CNS and produce a local response. On the other hand, short-chain fatty acids derived from the GI microbiota can inhibit amyloid aggregation [217][87]. Additionally, it seems feasible that GI-derived amyloid and toxins might activate signaling pathways affecting neuroinflammation and the pathogenesis of AD [218][88].
Furthermore, the association between misfolded proteins and cellular membrane damage is also modulated by the activation of membrane receptors that influence the neuroinflammatory response in the brain. For instance, the enhancement of inflammatory markers released from brain astrocytes is associated with AD and PD [219][89]. Additionally, it is believed that metabotropic glutamate receptor 5 (mGluR5) exerts an important action on neuroinflammation, affecting cytokine expression and activation of glial cells, such as microglia and astrocytes in the brain [219,220][89][90] (Figure 1). Activation of mGluR5 results in the stimulation of phospholipase C and phosphoinositide hydrolysis, leading to intracellular Ca2+ mobilization and activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) downstream signaling pathways, which might further affect neuroinflammation. mGluR5 activation contributes to a dysregulated rise in intracellular calcium concentration that is deleterious for neurons in AD and PD. For example, the exposure of neurons to Aβ oligomers induces mGluR5-dependent release of Ca2+ from the endoplasmic reticulum and toxicity [221,222][91][92]. This was corroborated using an mGluR5 knockout (KO), which showed reduced neutrophil infiltration and inflammatory cytokine expression in the brain at 24 h post-insult accompanied by improved neurological function [223][93]. In addition, mGluR5 KO showed reduced damage to BBB integrity and permeability, which might affect the influx of inflammatory modulators and peripheral cells into the brain. Interestingly, activation of these metabotropic receptors led to increases in intracellular calcium, further potentiating its increase due to direct membrane damage by these oligomeric toxic complexes.
References
- Alzheimer’s Disease International. World Alzheimer Report 2019: Attitudes to Dementia 2019.
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783.
- Stefanacci, R.G. The costs of Alzheimer’s disease and the value of effective therapies. Am. J. Manag. Care 2011, 17, S356.
- Alzheimer’s, A. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429.
- Panegyres, P.K.; Chen, H.Y. Coalition against Major, D. Early‐onset A lzheimer’s disease: A global cross‐sectional analysis. Eur. J. Neurol. 2014, 21, 1149-e65.
- Pierce, A.L.; Bullain, S.S.; Kawas, C.H. Late-onset Alzheimer disease. Neurol. Clin. 2017, 35, 283–293.
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Amyloid β oligomers (AβOs) in Alzheimer’s disease. J. Neural. Transm. 2017, 125, 177–191.
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791.
- MacLeod, R.; Hillert, E.-K.; Cameron, R.T.; Baillie, G.S. The role and therapeutic targeting of α-, β-and γ-secretase in Alzheimer’s disease. Future Sci. OA 2015, 1, doi:10.4155/fso.15.9.
- Fernandez-Perez, E.J.; Peters, C.; Aguayo, L.G. Membrane damage induced by amyloid beta and a potential link with neuroinflammation. Curr. Pharm. Des. 2016, 22, 1295–1304.
- Sepulveda, F.J.; Parodi, J.; Peoples, R.W.; Opazo, C.; Aguayo, L.G. Synaptotoxicity of Alzheimer beta amyloid can be explained by its membrane perforating property. PLoS ONE 2010, 5, e11820.
- Diaz, J.C.; Simakova, O.; Jacobson, K.A.; Arispe, N.; Pollard, H.B. Small molecule blockers of the Alzheimer Aβ calcium channel potently protect neurons from Aβ cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 3348–3353.
- Sepúlveda, F.J.; Fierro, H.; Fernandez, E.; Castillo, C.; Peoples, R.W.; Opazo, C.; Aguayo, L.G. Nature of the neurotoxic membrane actions of amyloid-β on hippocampal neurons in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 472–481.
- Killin, L.O.J.; Starr, J.M.; Shiue, I.J.; Russ, T.C. Environmental risk factors for dementia: A systematic review. BMC Geriatr. 2016, 16, 175, doi:10.1186/s12877-016-0342-y.
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; Collado-Mateo, D. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953.
- Tysnes, O.-B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural. Transm. 2017, 124, 901–905.
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 1–21.
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912.
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299, doi:10.3389/fnmol.2019.00299.
- Uttley, L.; Carroll, C.; Wong, R.; Hilton, D.A.; Stevenson, M. Creutzfeldt-Jakob disease: A systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect. Dis. 2020, 20, e2–e10.
- N.I.H. Creutzfeldt-Jakob Disease Fact Sheet.
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306, doi:10.1038/81834.
- Leemans, M. Prion diseases. Anaesth. Intensive Care Med. 2019, 21, 56–59.
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic proteins in neurodegenerative disease. Science 2002, 296, 1991–1995.
- Chung, C.G.; Lee, H.; Lee, S.B. Mechanisms of protein toxicity in neurodegenerative diseases. Cell. Mol. Life Sci. 2018, 75, 3159–3180.
- Hardy, J. A hundred years of Alzheimer’s disease research. Neuron 2006, 52, 3–13.
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 2007, 27, 2866–2875.
- Brindle, N.; George-Hyslop, P.S. The genetics of Alzheimer’s disease. Methods Mol. Med. 2000, 32, 23–43, doi:10.1385/1-59259-195-7:23.
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature 2006, 440, 352–357.
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842.
- Peters, C.; Fernandez-Perez, E.J.; Burgos, C.F.; Espinoza, M.P.; Castillo, C.; Urrutia, J.C.; Streltsov, V.A.; Opazo, C.; Aguayo, L.G. Inhibition of amyloid beta-induced synaptotoxicity by a pentapeptide derived from the glycine zipper region of the neurotoxic peptide. Neurobiol. Aging 2013, 34, 2805–2814.
- Liebert, A.; Bicknell, B.; Adams, R. Prion protein signaling in the nervous system—A review and perspective. Sign. Transduct. Insights 2014, 3, STI-S12319.
- Pacheco, C.R.; Morales, C.N.; Ramírez, A.E.; Muñoz, F.J.; Gallegos, S.S.; Caviedes, P.A.; Aguayo, L.G.; Opazo, C.M. Extracellular α-synuclein alters synaptic transmission in brain neurons by perforating the neuronal plasma membrane. J. Neurochem. 2015, 132, 731–741, doi:10.1111/jnc.13060.
- Ghio, S.; Camilleri, A.; Caruana, M.; Ruf, V.C.; Schmidt, F.; Leonov, A.; Ryazanov, S.; Griesinger, C.; Cauchi, R.J.; Kamp, F.; et al. Cardiolipin promotes pore-forming activity of alpha-synuclein oligomers in mitochondrial membranes. ACS Chem. Neurosci. 2019, 10, 3815–3829, doi:10.1021/acschemneuro.9b00320.
- Österlund, N.; Moons, R.; Ilag, L.L.; Sobott, F.; Graslund, A. Native ion mobility-mass spectrometry reveals the formation of β barrel shaped amyloid β hexamers in a membrane-mimicking environment. J. Am. Chem. Soc. 2019, 141, 10440–10450, doi:10.1021/jacs.9b04596.
- Chich, J.-F.; Chapuis, C.; Henry, C.; Vidic, J.; Rezaei, H.; Noinville, S. Vesicle permeabilization by purified soluble oligomers of prion protein: A comparative study of the interaction of oligomers and monomers with lipid membranes. J. Mol. Biol. 2010, 397, 1017–1030, doi:10.1016/j.jmb.2010.02.013.
- Combet, S.; Cousin, F.; Rezaei, H.; Noinville, S. Membrane interaction of off-pathway prion oligomers and lipid-induced on-pathway intermediates during prion conversion: A clue for neurotoxicity. Biochim. Biophys. Acta-Biomembr. 2019, 1861, 514–523, doi:10.1016/j.bbamem.2018.12.001.
- Pan, J.; Sahoo, P.K.; Dalzini, A.; Hayati, Z.; Aryal, C.M.; Teng, P.; Cai, J.; Gutierrez, H.R.; Song, L. Membrane disruption mechanism of a Prion Peptide (106-126) investigated by atomic force microscopy, raman and electron paramagnetic resonance spectroscopy. J. Phys. Chem. B 2017, 121, 5058–5071, doi:10.1021/acs.jpcb.7b02772.
- Paulis, D.; Maras, B.; Schinin, M.E.; Francesco, L. Di; Principe, S.; Galeno, R.; Abdel-Haq, H.; Cardone, F.; Florio, T.; Pocchiari, M.; et al. The pathological prion protein forms ionic conductance in lipid bilayer. Neurochem. Int. 2011, 59, 168–174, doi:10.1016/j.neuint.2011.04.008.
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219.
- Abu-Rumeileh, S.; Oeckl, P.; Baiardi, S.; Halbgebauer, S.; Steinacker, P.; Capellari, S.; Otto, M.; Parchi, P. CSF ubiquitin levels are higher in Alzheimer’s disease than in frontotemporal dementia and reflect the molecular subtype in prion disease. Biomolecules 2020, 10, 497.
- Tanaka, M.; Toldi, J.; Vecsei, L. Exploring the etiological links behind neurodegenerative diseases: Inflammatory cytokines and bioactive kynurenines. Int J. Mol. Sci. 2020, 21, 2431.
- Nelson, P.T.; Soma, L.A.; Lavi, E. Microglia in diseases of the central nervous system. Ann. Med. 2002, 34, 491–500.
- Liu, B.; Hong, J.S. Role of microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 2003, 304, 1–7.
- Garcao, P.; Oliveira, C.R.; Agostinho, P. Comparative study of microglia activation induced by amyloid-beta and prion peptides: Role in neurodegeneration. J. Neurosci. Res. 2006, 84, 182–193.
- Paranjape, G.S.; Gouwens, L.K.; Osborn, D.C.; Nichols, M.R. Isolated amyloid-beta(1-42) protofibrils, but not isolated fibrils, are robust stimulators of microglia. ACS Chem. Neurosci. 2012, 3, 302–311.
- Tu, J.; Chen, B.; Yang, L.; Qi, K.; Lu, J.; Zhao, D. Amyloid-beta activates microglia and regulates protein expression in a manner similar to prions. J. Mol. Neurosci. 2015, 56, 509–518.
- Heneka, M.T.; Carson, M.J.; Khoury, J. El; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405.
- Vom Berg, J.; Prokop, S.; Miller, K.R.; Obst, J.; Kälin, R.E.; Lopategui-Cabezas, I.; Wegner, A.; Mair, F.; Schipke, C.G.; Peters, O.; et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s diseasea-like pathology and cognitive decline. Nat. Med. 2012, 18, 1812–1819, doi:10.1038/nm.2965.
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19.
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542.
- Saitgareeva, A.R.; Bulygin, K.V.; Gareev, I.F.; Beylerli, O.A.; Akhmadeeva, L.R. The role of microglia in the development of neurodegeneration. Neurol. Sci. 2020, doi:10.1007/s10072-020-04468-5.
- Puoti, G.; Giaccone, G.; Mangieri, M.; Limido, L.; Fociani, P.; Zerbi, P.; Suardi, S.; Rossi, G.; Iussich, S.; Capobianco, R.; et al. Sporadic Creutzfeldt-Jakob disease: The extent of microglia activation is dependent on the biochemical type of PrPSc. J. Neuropathol. Exp. Neurol. 2005, 64, 902–909.
- Franceschini, A.; Strammiello, R.; Capellari, S.; Giese, A.; Parchi, P. Regional pattern of microgliosis in sporadic Creutzfeldt-Jakob disease in relation to phenotypic variants and disease progression. Neuropathol. Appl. Neurobiol. 2018, 44, 574–589.
- Makarava, N.; Chang, J.C.; Molesworth, K.; Baskakov, I. V Region-specific glial homeostatic signature in prion diseases is replaced by a uniform neuroinflammation signature, common for brain regions and prion strains with different cell tropism. Neurobiol. Dis. 2020, 137, 104783.
- Aguzzi, A.; Liu, Y. A role for astroglia in prion diseases. J. Exp. Med. 2017, 214, 3477–3479.
- Aguzzi, A.; Zhu, C. Microglia in prion diseases. J. Clin. Invest. 2017, 127, 3230–3239.
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194.
- Taipa, R.; das Neves, S.P.; Sousa, A.L.; Fernandes, J.; Pinto, C.; Correia, A.P.; Santos, E.; Pinto, P.S.; Carneiro, P.; Costa, P.; et al. Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol. Aging 2019, 76, 125–132, doi:10.1016/j.neurobiolaging.2018.12.019.
- Stoeck, K.; Bodemer, M.; Ciesielczyk, B.; Meissner, B.; Bartl, M.; Heinemann, U.; Zerr, I. Interleukin 4 and interleukin 10 levels are elevated in the cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. Arch. Neurol. 2005, 62, 1591–1594.
- Stoeck, K.; Bodemer, M.; Zerr, I. Pro- and anti-inflammatory cytokines in the CSF of patients with Creutzfeldt-Jakob disease. J. Neuroimmunol. 2006, 172, 175–181.
- Garcia-Revilla, J.; Alonso-Bellido, I.M.; Burguillos, M.A.; Herrera, A.J.; Espinosa-Oliva, A.M.; Ruiz, R.; Cruz-Hernandez, L.; Garcia-Dominguez, I.; Roca-Ceballos, M.A.; Santiago, M.; et al. Reformulating pro-oxidant microglia in neurodegeneration. J. Clin. Med. 2019, 8, 1719.
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337.
- Zhou, S.L.; Tan, C.C.; Hou, X.H.; Cao, X.P.; Tan, L.; Yu, J.T. TREM2 variants and neurodegenerative diseases: A systematic review and meta-analysis. J. Alzheimers Dis. 2019, 68, 1171–1184.
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Pina-Crespo, J.C.; Zhang, M.; et al. TREM2 is a receptor for beta-amyloid that mediates microglial function. Neuron 2018, 97, 1023–1031.e7.
- Zhong, L.; Wang, Z.; Wang, D.; Wang, Z.; Martens, Y.A.; Wu, L.; Xu, Y.; Wang, K.; Li, J.; Huang, R.; et al. Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol. Neurodegener. 2018, 13, 15.
- Jana, M.; Palencia, C.A.; Pahan, K. Fibrillar amyloid-beta peptides activate microglia via TLR2: Implications for Alzheimer’s disease. J. Immunol. 2008, 181, 7254–7262.
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562.
- Lukiw, W.J. Gastrointestinal (GI) tract microbiome-derived neurotoxins-potent neuro-inflammatory signals from the GI tract via the systemic circulation into the brain. Front. Cell Infect. Microbiol. 2020, 10, 22.
- Daws, M.R.; Sullam, P.M.; Niemi, E.C.; Chen, T.T.; Tchao, N.K.; Seaman, W.E. Pattern recognition by TREM-2: Binding of anionic ligands. J. Immunol. 2003, 171, 594–599.
- Batista, I.A.; Melo, S.A. Exosomes and the future of immunotherapy in pancreatic cancer. Int. J. Mol. Sci. 2019, 20, 567.
- Cunningham, C.; Campion, S.; Lunnon, K.; Murray, C.L.; Woods, J.F.; Deacon, R.M.; Rawlins, J.N.; Perry, V.H. Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol. Psychiatry 2009, 65, 304–312.
- Sama, D.M.; Norris, C.M. Calcium dysregulation and neuroinflammation: Discrete and integrated mechanisms for age-related synaptic dysfunction. Ageing Res. Rev. 2013, 12, 982–995.
- Sender, R.; Fuchs, S.; Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016, 14, e1002533.
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Heal. Dis. 2015, 26, doi:10.3402/mehd.v26.26191.
- Wilkins, L.J.; Monga, M.; Miller, A.W. Defining dysbiosis for a cluster of chronic diseases. Sci. Rep. 2019, 9, 12918, doi:10.1038/s41598-019-49452-y.
- Erickson, M.A.; Dohi, K.; Banks, W.A. Neuroinflammation: A common pathway in CNS diseases as mediated at the blood-brain barrier. Neuroimmunomodulation 2012, 19, 121–130, doi:10.1159/000330247.
- Sharma, K.; Kalakoti, P.; Nanda, A.; Sun, H. Blood-brain barrier disruption during neuroinflammation. In Neuroinflammation; Elsevier: Amsterdam, The Netherlands, 2018; pp. 529–539.
- Bhattacharjee, S.; Lukiw, W.J. Alzheimer’s disease and the microbiome. Front. Cell. Neurosci. 2013, 7, 153, doi:10.3389/fncel.2013.00153.
- Asti, A.; Gioglio, L. Can a bacterial endotoxin be a key factor in the kinetics of amyloid fibril formation? J. Alzheimer’s Dis. 2014, 39, 169–179, doi:10.3233/JAD-131394.
- Lee, J.; Lee, Y.; Yuk, D.; Choi, D.; Ban, S.; Oh, K.; Hong, J. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflamm. 2008, 5, 37, doi:10.1186/1742-2094-5-37.
- Blasko, I.; Marx, F.; Steiner, E.; Hartmann, T.; Grubeck‐Loebenstein, B. TNFα plus IFNγ induce the production of Alzheimer β‐amyloid peptides and decrease the secretion of APPs. FASEB J. 1999, 13, 63–68, doi:10.1096/fasebj.13.1.63.
- Buxbaum, J.D.; Oishi, M.; Chen, H.I.; Pinkas-Kramarski, R.; Jaffe, E.A.; Gandy, S.E.; Greengard, P. Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer β/A4 amyloid protein precursor. Proc. Natl. Acad. Sci. USA 1992, 89, 10075–10078, doi:10.1073/pnas.89.21.10075.
- Evans, M.L.; Chorell, E.; Taylor, J.D.; Åden, J.; Götheson, A.; Li, F.; Koch, M.; Sefer, L.; Matthews, S.J.; Wittung-Stafshede, P.; et al. The bacterial curli system possesses a potent and selective inhibitor of amyloid formation. Mol. Cell 2015, 57, 445–455, doi:10.1016/j.molcel.2014.12.025.
- Chen, S.G.; Stribinskis, V.; Rane, M.J.; Demuth, D.R.; Gozal, E.; Roberts, A.M.; Jagadapillai, R.; Liu, R.; Choe, K.; Shivakumar, B.; et al. Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged fischer 344 rats and caenorhabditis elegans. Sci. Rep. 2016, 6, 1–10, doi:10.1038/srep34477.
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802, doi:10.1038/srep41802.
- Ho, L.; Ono, K.; Tsuji, M.; Mazzola, P.; Singh, R.; Pasinetti, G.M. Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert Rev. Neurother. 2018, 18, 83–90, doi:10.1080/14737175.2018.1400909.
- Pistollato, F.; Cano, S.S.; Elio, I.; Vergara, M.M.; Giampieri, F.; Battino, M. Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr. Rev. 2016, 74, 624–634, doi:10.1093/nutrit/nuw023.
- Shah, A.; Silverstein, P.S.; Singh, D.P.; Kumar, A. Involvement of metabotropic glutamate receptor 5, AKT/PI3K Signaling and NF-κB pathway in methamphetamine-mediated increase in IL-6 and IL-8 expression in astrocytes. J. Neuroinflamm. 2012, 9, 547, doi:10.1186/1742-2094-9-52.
- Chantong, B.; Kratschmar, D.V.; Lister, A.; Odermatt, A. Inhibition of metabotropic glutamate receptor 5 induces cellular stress through pertussis toxinsensitive Gi-proteins in murine BV-2 microglia cells. J. Neuroinflamm. 2014, 11, 190, doi:10.1186/s12974-014-0190-7.
- Renner, M.; Lacor, P.N.; Velasco, P.T.; Xu, J.; Contractor, A.; Klein, W.L.; Triller, A. Deleterious effects of amyloid β oligomers acting as an extracellular scaffold for mGluR5. Neuron 2010, 66, 739–754, doi:10.1016/j.neuron.2010.04.029.
- Shrivastava, A.N.; Aperia, A.; Melki, R.; Triller, A. Physico-pathologic mechanisms involved in neurodegeneration: Misfolded protein-plasma membrane interactions. Neuron 2017, 95, 33–50.
- Yang, T.; Liu, Y.W.; Zhao, L.; Wang, H.; Yang, N.; Dai, S.S.; He, F. Metabotropic glutamate receptor 5 deficiency inhibits neutrophil infiltration after traumatic brain injury in mice. Sci. Rep. 2017, 7, 1–12, doi:10.1038/s41598-017-10201-8.