Despite pharmacological treatments and surgical practice options, the mortality rate of astrocytomas and glioblastomas remains high, thus representing a medical emergency for which it is necessary to find new therapeutic strategies. Fibroblast growth factors (FGFs) act through their associated receptors (FGFRs), a family of tyrosine kinase receptors consisting of four members (FGFR1–4), regulators of tissue development and repair. In particular, FGFRs play an important role in cell proliferation, survival, and migration, as well as angiogenesis, thus their gene alteration is certainly related to the development of the most common diseases, including cancer. FGFRs are subjected to multiple somatic aberrations such as chromosomal amplification of FGFR1; mutations and multiple dysregulations of FGFR2; and mutations, translocations, and significant amplifications of FGFR3 and FGFR4 that correlate to oncogenesis process. Therefore, the in-depth study of these receptor systems could help to understand the etiology of both astrocytoma and glioblastoma so as to achieve notable advances in more effective target therapies. Furthermore, the discovery of FGFR inhibitors revealed how these biological compounds improve the neoplastic condition by demonstrating efficacy and safety.
- brain tumors
- astrocytoma
- glioblastoma
- fibroblas
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
In the context of central nervous system (CNS) disorders, tumors are certainly one of the most widespread and lethal pathologies. These types of cancers affect the CNS in all its parts: brain, bone marrow, and cerebellum (as summarized in Figure 1). Brain tumors are classified into two categories: primary tumors, which originate and develop directly in the central nervous tissue, and secondary tumors, or metastases, which derivate from tumors cells present in other organs, such as lung, breast, gastrointestinal tract, etc. and subsequently spread to nerve tissue [1][2][3][1–3].
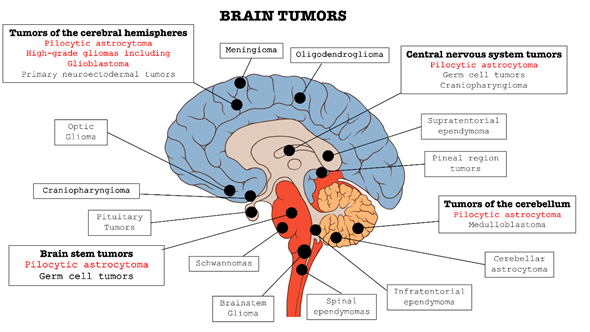
Figure 1. Classification of the main brain tumors; the tumors involved in this review are highlighted in red.
In the last years, there has been a progressive upsurge in brain tumors incidence; in particular, this increase was most significant in the over 65 age group, and higher in men than women [4]. The manifestations of a brain tumor depend mainly on its location and size [4] that can lead to compression, infiltration of healthy tissue, increasing intracranial pressure, and cerebral edema [5][6][5,6].
The onset of symptoms is usually insidious, may not be specific, and include headache, nausea, vomit, mental changes, balance disorders, speech disorders, lack of strength of the limbs, or sensitivity disorders [7][8][9][7–9], while an acute vascular complication, e.g. stroke, is a less frequent manifestation [10].
Pharmacological therapy changes considerably according to the age of the patient and the site of the neoplasm [11][12][11,12].
Chemotherapy may be useful in some types of brain tumors even if this therapeutic approach is hampered by the presence of the blood–brain barrier (BBB) [13][14][13,14]. BBB acts as a kind of natural filter that limits the passage of substances, and therefore also drugs, from the blood to the brain tissues [15]. Among the drugs capable of crossing BBB, the most used are alkylating agents such as Temozolomide and drugs belonging to the Nitrosoureas family such as Lomustine and Carmustine [16][17][16,17]. Often, more effective results have been obtained by combining drug therapy with radiotherapy [18]. In fact, the combination of the three drugs Lomustine, Procarbazine, and Vincristine [19] with radiotherapy contributed to significantly prolong the survival of patients with low-grade gliomas [20][21][20,21]. Over the years, radiotherapy (RT) has increasingly assumed a fundamental role in the treatment of primary brain tumors as well as metastases [22]. Indeed, thanks to advances in imaging and radiotherapy techniques, it has been possible to make a more precise localization of the tumor, thus allowing a reduction in the volume of irradiated healthy brain tissue [22]. All this led to a reduction in long-term toxicity due to radiotherapy, but with the same results in terms of efficacy.
In addition to these, surgical treatment represents one of the most valid options in the case of both primary tumors and metastases [23][24][23,24]. Moreover, the process of carcinogenesis in the brain involve various pathways and molecular mechanisms, many of which are still under study and deepening. Recent articles, for example, highlight the role of receptors such as interleukin 13 receptor alpha 2 (IL-13RA2) as a tumor-associated receptor over-expressed in most patients with glioblastoma as well as the overexpression of EphA2 and EphA3 receptors [25].
2. Role of FGFRs in Brain Tumors
Primary CNS tumors include a diverse set of pathological entities; however, despite the above-mentioned generic classification, it is more appropriate to distinguish brain tumors on the basis of their origin; a first distinction can be made by considering “non-glial tumors” and “glial tumors”.
These last types of tumors originate from glial cells and represent the most common form of all CNS tumors, representing 81% of malignant brain tumors [26][103]. They are classified into various subtypes, distinguished according to the cell type from which they originate and the degree of differentiation and/or malignancy [27][28][104,105]. Among the most common gliomas, we have astrocytomas (originating from astrocytic cells) including glioblastoma, oligodendrogliomas (from oligodendroglial cells), and ependymomas (from ependymal cells) [29][106].
Given that FGFRs play a significant role in cell proliferation, survival, and migration, as well as angiogenesis, their gene alteration is certainly correlated with the development of most common pathologies, including several types of cancer such as breast, bladder (specifically, urothelial cell carcinoma), lung (small cell length carcinoma and non-small cell lung carcinoma), prostate, and multiple myeloma [30][107]. In this review, we focus on their involvement in brain tumors, with particular attention on astrocytoma and glioblastoma, as summarized in Table 1.
Table 1. The subtypes of FGFRs (FGFR1–4) with related molecular weight, residues AA, malignancy type, and inhibitor FGFRs are shown. In addition, i a framework of the common FGFR alterations in brain tumors is also provided. The main mutations of FGFRs, for pilocytic astrocytoma and glioblastoma, are illustrated in the last two columns.
|
FGFRs |
Molecular Weight |
Residues AA |
Malignancy Type |
Inhibitor FGFRs |
Common FGFR Alterations in Brain Tumors |
Mutation of FGFRs in Pilocytic Astrocytoma |
Mutation of FGFRs in Glioblastoma |
|
FGFR1 |
91.9 kDa [31] |
822 [31] |
Glioblastomas Low grade brain gliomas [31] |
Futibatinib, Infigratinib AZD4547 [31] |
FGFR1-TKD [32] FGFR1-TACC1 Fusion [32] FGFR1 hotspot mutations: p.N546, p.K656 [32] |
Residues in αA1: N546K (KD1) [33] Residues in αB1: N544K (KD1) [33] Residues in αA1: K655I (KD2) [33] Residues in αB1: K653I (KD2) [33] Residues in αA1: K656D/E/M/N (KD2) [33] Residues in αB1: K654D/E/M/N (KD2) [33] Residues in αA1: T658P (KD2) [33] Residues in αB1: T656P (KD2) [33] |
Residues in αA1: N546K (KD1) [34] Residues in αB1: N544K (KD1) [34] Residues in αA1: R576W (KD1) [34] Residues in αB1: R574W (KD1) [34] Residues in αA1: K656E (KD2) [35] Residues in αB1: K654E (KD2) [35] |
|
FGFR2 |
92.0 kDa [31] |
821/822 [31] |
Glioblastomas, Low grade brain gliomas [31] |
Futibatinib, Infigratinib, AZD4547 [31] |
FGFR2-CTNNA3 fusion [32] |
Residues in IIIb: K660E (KD2) [33] Residues in IIIc: K659E (KD2) [33] |
Residues in IIIb: Q212K (IgII) [35] Residues in IIIc: Q212K (IgII) [35] Residues in IIIb: G463E (JM) [34] Residues in IIIc: G462E (JM) [34] |
|
FGFR3 |
87.7-88.2 kDa [31] |
806/808 [31] |
Glioblastomas, Low grade brain gliomas [31] |
Futibatinib, Infigratinib, AZD4547 [31] |
FGFR3-TACC3 fusions [32] |
Residues in IIIb: E468K (JM) [36] Residues in IIIc: E466K (JM) [36] Residues in IIIb: R605Q (KD2) [37] Residues in IIIc: R603Q (KD2) [37] |
|
|
FGF4 |
88.0 kDa [31] |
802 [31] |
Glioblastomas, Low grade brain gliomas [31] |
Fisogatinib [31] |
Residues in P22455-1: Q144E (IgI – IgII) [35] Residues in P22455-2: Q144E (IgI – IgII) [35] Residues in P22455-1: R434Q (JM) [35] Residues in P22455-2: R394Q (JM) [35] |
||
|
FGFRs |
Molecular Weight |
Residues AA |
Malignancy Type |
Inhibitor FGFRs |
Common FGFR Alterations in Brain Tumors |
Mutation of FGFRs in Pilocytic Astrocytoma |
Mutation of FGFRs in Glioblastoma |
|
FGFR1 |
91.9 kDa [37] |
822 [37] |
Glioblastomas Low grade brain gliomas [37] |
Futibatinib, Infigratinib AZD4547 [37] |
FGFR1-TKD [80] FGFR1-TACC1 Fusion [80] FGFR1 hotspot mutations: p.N546, p.K656 [80] |
Residues in αA1: N546K (KD1) [108] Residues in αB1: N544K (KD1) [108] Residues in αA1: K655I (KD2) [108] Residues in αB1: K653I (KD2) [108] Residues in αA1: K656D/E/M/N (KD2) [108] Residues in αB1: K654D/E/M/N (KD2) [108] Residues in αA1: T658P (KD2) [108] Residues in αB1: T656P (KD2) [108] |
Residues in αA1: N546K (KD1) [81] Residues in αB1: N544K (KD1) [81] Residues in αA1: R576W (KD1) [81] Residues in αB1: R574W (KD1) [81] Residues in αA1: K656E (KD2) [109] Residues in αB1: K654E (KD2) [109] |
|
FGFR2 |
92.0 kDa [37] |
821/822 [37] |
Glioblastomas, Low grade brain gliomas [37] |
Futibatinib, Infigratinib, AZD4547 [37] |
FGFR2-CTNNA3 fusion [80] |
Residues in IIIb: K660E (KD2) [108] Residues in IIIc: K659E (KD2) [108] |
Residues in IIIb: Q212K (IgII) [109] Residues in IIIc: Q212K (IgII) [109] Residues in IIIb: G463E (JM) [82] Residues in IIIc: G462E (JM) [82] |
|
FGFR3 |
87.7-88.2 kDa [37] |
806/808 [37] |
Glioblastomas, Low grade brain gliomas [37] |
Futibatinib, Infigratinib, AZD4547 [37] |
FGFR3-TACC3 fusions [80] |
Residues in IIIb: E468K (JM) [110] Residues in IIIc: E466K (JM) [110] Residues in IIIb: R605Q (KD2) [111] Residues in IIIc: R603Q (KD2) [111] |
|
|
FGF4 |
88.0 kDa [37] |
802 [37] |
Glioblastomas, Low grade brain gliomas [37] |
Fisogatinib [37] |
Residues in P22455-1: Q144E (IgI – IgII) [109] Residues in P22455-2: Q144E (IgI – IgII) [109] Residues in P22455-1: R434Q (JM) [109] Residues in P22455-2: R394Q (JM) [109] |
2.1. Role of FGFRs in Astrocytoma
Astrocytoma is a tumor that originates from a specific type of glial cell: “astrocytes” [38][112]. Astrocytes are the most differentiated glial cells, characterized by elaborate radially symmetrical branches, which attribute to them the characteristic star shape [39][113]. On the basis of their geometry, they are distinguished into protoplasmic forms, when short and rare extensions are present; fibrous forms, when they have numerous long and thin cytoplasmic; or radial extensions, of elongated shape and distributed perpendicular to the axis of the ventricles [40][114]. Astrocytomas represent the most common forms of gliomas [41][115], representing 80% of malignant tumors of CNS [42][116]; specifically, low-grade gliomas are more frequent in young ages [43][44][117,118], while anaplastic or malignant gliomas generally have a later onset [45][119], even if there are exceptions.
There are several classifications proposed, in general low or high malignancy astrocytomas are distinguished while in particular a distinction is made based on four degrees: I, II, III, and IV [46][120].
The main categories of astrocytomas are: pilocytic astrocytoma and subependymal giant cell (grade I), diffuse astrocytomas (grade II), pleomorphic xanthastrocytomas (grades II and III), anaplastic astrocytoma (grade III), and glioblastoma (grade IV) [47][121]. These various degrees of histological variability correspond to various degrees of malignancy, which is given both by the rapidity of growth and by the ability to reform themselves after surgical removal [104].
Therefore, the assessment of gradation is an important parameter for both prognosis and therapy [104].
The latest revision proposed by the World Health Organization (WHO) classifies gliomas by integrating the data of conventional histological analysis with molecular information obtained through specific genetic analyses [48][122]. In particular, it is highlighted how the presence or absence of a mutation in the isocitrate dehydrogenase (IDH) 1/2 gene [49][50][51][123–125], deletion of chromosome arms 1p and 19q (1p/19q codeletion), and mutations in the TERT promoter are determining factors in establishing a specific histomolecular subtype [52][53][126,127].
The causes that lead to the formation and development of astrocytoma are still little known and in continuous analysis. However, it is recognized that defects related to chromosomal and genetic mutations play a decisive role in the uncontrolled growth of brain cells, involving multiple mechanisms and pathways, in which FGFRs also contribute [108].
This statement is confirmed in a study by Lew et al. [54][128], in which the oncogenic role played by FGFRs receptors, and in particular by FGFR1, is well highlighted. Specifically, the precise sequence of FGF receptor autophosphorylation is kinetically driven and is disrupted by oncogenic mutations. Such involvement of FGFRs in oncogenesis processes made these receptors one of the most promising targets for FGFR-derived cancer therapies. Thus, drugs targeting FGFRs could be an effective therapeutic approach for cancers [55][129].
Moreover, a study by Sie et al. [56][130] proved how pediatric low-grade astrocytoma cell line showed high percentages of cells expressing FGFR1 (34–51%) compared to isotype controls. The study showed how the inhibition of FGFR1 decreased tumor cell viability, thus highlighting the importance of environmental growth factors in developing tumor escape towards RTK inhibitors [56][130]. Similar results were reported by Trisolini et al. [57][131], which highlight the frequent FGFR1 mutation in optic-pathway pilocytic astrocytomas, revealing FGFR1 as an excellent candidate for anti-FGFR therapies in patients [57][131].
The involvement of FGFR1 and FGFR3 in pilocytic astrocytoma was also confirmed by Lehtinen et al. [58][132]. In this study, immunohistochemical analysis revealed how moderate-to-strong FGFR3 expression was detected predominantly in non-pediatric patients [58][132]. In addition to this, strong expression of the FGFR3 protein is indicative of FGFR3 fusions and may serve as a clinically applicable predictive marker for FGFR inhibitor-based treatment regimens [59][133].
Further evaluations on FGFR3 and in particular on gene fusions of FGFR3-TACC3 (F3-T3) were carried out by Frattini et al. [60][134], elucidating the oncogenic circuitry activated by F3-T3, showing that F3-T3 positive tumors rely on mitochondrial respiration, and highlighting this pathway as a therapeutic opportunity for the treatment of tumors with F3-T3 fusions [60][134].
FGFR-TACC fusions generate potent oncogenes that combine growth-promoting effects with aneuploidy through the activation of still unclear intracellular signaling mechanisms [61][135]. In relation to this, clinical data show promising effects in cancer patients hosting FGFR-TACC fusions and treated with FGFR inhibitors [61][135], therefore future insights could lead to encouraging results.
2.2. Role of FGFRs in Glioblastoma
Gliomas are malignant primary brain tumors, among which glioblastoma has the worst prognosis [62][136]. According to the most recent WHO guidelines, it is classified as a grade IV diffuse astrocytoma [62][136]. Glioblastoma is a particularly aggressive type of cancer affecting the glial cells, in particular astrocytes, which have a supporting role in CNS [63][137]. Furthermore, it represents about 45.2% of all malignant CNS tumors [64][65][138,139] with an incidence of 5–6 cases per 100,000 people [66][140].
Glioblastoma is characterized by uncontrolled proliferation, angiogenesis, invasiveness, and necrosis [67][141], and it can develop de novo or through the malignant progression of lower-grade astrocytomas [68][142]. Numerous risk factors leading to the development of glioblastoma have been identified as genetic factors and environmental factors including exposure to therapeutic ionizing radiation, pesticides, and smoking [66][140]. Glioblastoma is usually described in two different clinical forms, primary and secondary; primary glioblastoma is the most common form (about 95%) and arises typically de novo, within 3–6 months, in older patients, while secondary glioblastoma arises from prior low-grade astrocytomas (over 10–15 years) in younger patients [68][142]. The majority of glioblastoma tumors occur in the frontal lobes of the supratentorial compartments, in particular the temporal and parietal lobes, but they also occur in the cerebellum and spinal cord. In the last decade, many studies have been conducted to understand the role of genetic mutations and microenvironment in glioblastoma tumorigenesis [69][109–111,143].
Glioblastoma exhibits several cytogenetic abnormalities involving the loss or structural rearrangement of loci on chromosomes 9, 10, and 17 [70][71][72][144–146]. Many studies have revealed that somatic mutations of FGFRs signaling are among the most frequent molecular alterations that occur in glioblastoma, being involved in progression and growth tumor [73][74][147,148]. Deregulation of FGFR signaling is frequently observed in many types of cancer including glioblastoma, promoting the development and growth of cancers cell [75][149]. Gene expression analysis revealed profound heterogeneity of FGFR1–4 expression in glioblastoma patients [73][147]. Altered FGFR expression in astrocytes can lead to glioblastoma progression due to the activation of mitogenic, migratory, and antiapoptotic responses [73][147]. Several studies have reported that FGFR1 and FGFR2 gene amplification, abnormal activation, or single nucleotide polymorphisms (SNPs) have a key role in glioblastoma progression [73][135,147]. In this context, it has been shown that FGFR1 expression increases as the tumor progresses from benign to malignant, whereas FGFR2 levels in human gliomas gradually diminish [76][150]. Moreover, a recent report found that FGFR3 and FGFR4 are also expressed in invasive glioblastoma cells [98]. Scientific evidence reveals that human glioblastoma is also characterized by oncogenic fusions involving the members of the FGFR3 and FGFR1 tyrosine kinases (TKs) to the transforming acidic coiled-coil (TACC) proteins, in particular TACC3 and TACC1 [135], necessary to promote cell division [77][149,151]. The fusion between FGFR3 and TACC3 genes generates an oncogenic FGFR3 form [75][149]. It was reported that the ectopic expression of FGFR3-TACC3 fusion affects about 3% of glioblastoma patients, and its activation through dimerization and transphosphorylation of kinase domain contribute to carcinogenic events closely related to glioblastoma progression [135,152].
Furthermore, it is important to highlight that the mechanism of autocrine stimulation, in the context of glioblastoma, contributes to cell growth and invasion.
The high invasiveness of tumor cells remains one of the most critical challenges in the clinical management of patients with glioma [79][153] and in particular for glioblastoma patients.
This invasion of glioma cells is stimulated by both autocrine and paracrine factors which act on a wide range of cell surface-bound receptors. Among the key signaling elements that mediate receptor-initiated signaling in regulating glioblastoma invasion, there are Rho family GTPases [80][154], but FGFs play also their role, as in the case of basic fibroblast growth factor (FGF2, also called bFGF) [81][82][83][155–157].
This process of self-renew and tumor proliferation of glioblastoma cells by FGFs were also described by Allerstorfer et al. [84][158]. They demonstrated the contribution of FGF5 in the malignant progression of human astrocytic brain tumors by both autocrine and paracrine mechanisms. Moreover, their data indicate FGF5 exerts oncogenic activities in astrocytic brain tumors by promoting growth, survival, and migration effects on tumor cells and by supporting neoangiogenic processes [84][158]. siRNA-mediated FGF5 downregulation thus leads to a significant reduction in glioblastoma cell proliferation [84][158]; therefore, the silencing of this factor represents a promising target for therapeutic interventions in human glioblastoma.
Accordingly, a greater understanding of the molecular mechanisms that control invasion of glioblastoma cells may lead to the identification of future molecular targets for therapeutic intervention in this devastating disease.