Prenatal detection of uniparental disomy (UPD) is a methodological challenge, and a positive testing result requires comprehensive considerations on the clinical consequences as well as ethical issues.
- uniparental disomy,prenatal testing,next generation genomics
1. Introduction
Chromosomal abnormalities significantly contribute to reproductive failure and congenital disorders in humans, and meiotic errors resulting in aneuploidy account for the majority of first trimester pregnancy losses. A result of meiotic or mitotic chromosomal malsegregation is uniparental disomy (UPD) (Figure 1), the exceptional inheritance of the two chromosomes of a pair from the same parent [1], which in contrast to numeric or structural chromosomal aberration, does not affect number and structure of chromosomes. UPD therefore escapes cytogenetic detection, but with the increasing application of high resolution and throughput analysis in diagnostic genetic testing it is detected.
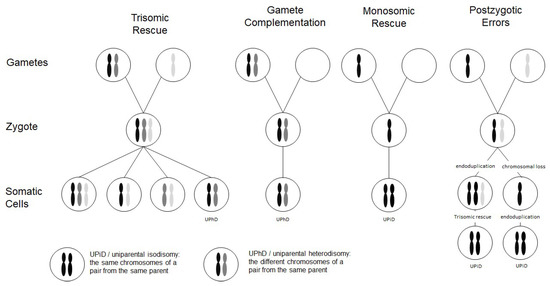
Figure 1. Scheme depicting the four major formation mechanisms of whole chromosome uniparental disomies (UPDs). As it can be delineated from these modes of formation, mosaicism of a UPD cell line as well as an aneuploidy cell line is possible in case of trisomic rescue and postzygotic errors (for further details see [2]). It should be noted that in case of monosomic rescue and postzygotic nondisjunction, uniparental idisomy of the whole chromosome can be observed, whereas in the other modes of formation, uniparental heterodisomy can be interrupted by stretches of uniparental disomy. Formation of segmental UPD as well as UPDs caused by structural chromosomal variants (like Robertsonian translocation) are not shown (for that issue see [3]).
UPD has meanwhile been reported for nearly all human chromosomes. In prenatal diagnosis it can be detected as an incidental finding in the course of genetic tests based on undirected approaches like genome-wide assays. These tests comprise microarray-based molecular karyotyping methods and next generation sequencing (NGS) approaches, and they are increasingly implemented in prenatal genetic testing. In fact, in case of UPD detection, the decision on the further management of the pregnancy requires considerations on methodological aspects, clinical and ethical issues.
2. Formation Mechanisms of UPD
Two types of UPD can be discriminated: in uniparental heterodisomy (UPhD), the cell has inherited the two different chromosomes from the same parent, whereas in uniparental isodisomy (UPiD) the same chromosome has been transmitted twice (Figure 1). In fact, complete UPhD or UPiD affecting a whole chromosome can be observed, but frequently stretches of UPhD alternate with UPiD regions on the affected chromosome (mixed UPhD/UPiD).
A major exception is paternal UPD 11 in Beckwith–Wiedemann syndrome (BWS) (Table 1) which is exclusively UPiD. UPD does not always affect a whole chromosome, but also segmental UPDs (also called partial UPD) comprising only segments of a chromosome have been reported.
UPD can occur after meiotic nondisjunctional errors, but mitotic formation mechanisms have been identified as well (Figure 1):
- The major formation mechanism is trisomic rescue, with the subsequent loss of one of the supernumerary chromosomes as a prerequisite for the cell to survive. In one third of these reductions, the chromosome from the parent contributing only one chromosome to the trisomic rescue is lost, thus giving rise to a UPD.
- Gamete complementation describes the fertilization of a disomic gamete by a nullisomic gamete, and is therefore also the result of meiotic errors. The result is complete UPhD.
- Survival of a monosomic gamete is possible by monosomic rescue via endoduplication, resulting in complete UPiD.
- Postfertilization UPD formation consists of mitotic malsegregations, either causing a monosomic cell with subsequent endoduplication, or an endoduplication followed by a chromosomal loss. In any case, postzygotic UPD is a complete UPiD.
Depending on the time of meiotic or mitotic nondisjunction, UPDs can occur as a mosaic constitution, even in combination with trisomic cell lines (Figure 1). Accordingly, detection of UPDs can be significantly hampered by the co-occurrence of chromosomal mosaicism, and UPDs might therefore escape detection.
3. Risk Factors Predisposing for UPD Formation
By trisomic rescue, every human chromosome can be affected by UPD, and UPD has meanwhile been reported for nearly all human chromosomes (for review: Liehr T. 2020. Cases with uniparental disomy http://cs-tl.de/DB/CA/UPD/0-Start.html (accessed 11 May 2020)). Accordingly, the frequency of single UPDs correlates with that of the respective trisomies in humans. In fact, trisomy 16 is the most common autosomal trisomy in humans [4][22] and nearly all trisomy 16 pregnancies originate from maternal meiosis errors [5][23]. It is therefore not surprising that maternal UPD of chromosome 16 (upd(16)mat) occurs relatively frequently (for review: [6][24]).
The link between trisomy and UPD is also reflected by increased maternal age in UPhD in comparison to UPiD (e.g., for upd(7)mat [7][25]), as increased maternal age is a relevant risk factor for trisomy formation, and so it is for UPD formation.
Up to 65% of UPDs are detected in individuals with a normal karyotype (46, XX; 46, XY) [8][26], and the others are associated with chromosomal rearrangements. As these variants predispose to an improper chromosomal segregation in meiosis and mitosis, they can be regarded as another risk factor for UPD formation. In principle, nearly every type of rearrangement can cause a UPD (for review: [9][27]), but in particular in families transmitting Robertsonian translocations (RobTk) are at risk for UPD formation. RobTks result from whole arm rearrangements of the five acrocentric chromosomes (chromosomes 13, 14, 15, 21, 22) and have an incidence of 1 in 1,000 in the general population (for review: [10][28]). Due to this frequency and the increased risk of RobTk carriers for trisomy and subsequent UPD formation, in particular, UPDs of the clinically relevant chromosomes 14 and 15 are constantly identified in routine genetic testing. Another group of structural variants prone to UPD formation are small supernumerary isochromosomes, but these are not restricted to specific chromosomes [11][29].
4. Conclusions and Outlook
The implementation of next generation genomic assays with a considerable high-resolution (e.g., microarrays, next and third generation sequencing, genome imaging) in the genetic diagnostic setting has already been conducted, and numerous studies and publications have proven the power, appropriateness and reliability of the different methods to identify SNVs and copy number variants (CNVs) as well as epigenetic alterations. In addition, they allow the identification of UPD, thereby the functional and clinical relevance of the whole chromosome as well as segmental UPDs can be elucidated. In parallel to the validation of next generation genomic assays in postnatal genetic diagnostics, their application in prenatal management has been pursued. In fact, prenatal chromosome microarray analysis [12][44] and NIPT are meanwhile widely used screening tools, and prenatal NGS to identify monogenetic causes of fetal malformations is in establishment [13][47]. With the increased application of these approaches as first-line tests, the number of incidental findings will further increase, and these findings comprise UPDs as well. However, as shown in this review, the incidental identification of UPD is often difficult to manage, and should therefore be embedded in a comprehensive multidisciplinary management approach, in particular, it should include a high-quality ultrasound and clinical assistance. It must be embedded in genetic counselling, and ethical issues have to be addressed so that the patients can finally come to a self-determined decision.
Generally, pre-test counselling is also necessary to inform on the benefits, limitations and diagnostic scope of genetic tests.