In recent years, intensive research has focused on understanding the biochemical and molecular alterations contributing to chronic pain. One promising avenue of exploration centers around the brain-derived neurotrophic factor (
BDNF) gene, which influences circulating levels of BDNF, and has been implicated in initiating and/or perpetuating neuronal hyperexcitability, maladaptive neuroplasticity, and disinhibition at different levels of nociceptive pathways
[15].
BDNF assumes a central role in promoting brain homeostasis and neuronal survival, and also serves as a critical regulator of synaptic plasticity
[16]. Despite its crucial role in maintaining normal physiological functions, a less positive perspective emerges concerning chronic pain, where mounting evidence suggests a pro-nociceptive role for
BDNF [17][18][17,18]. Consequently,
BDNF is increasingly acknowledged as a pivotal perpetuating factor in chronic pain.
2. The Physiological Role of BDNF
Since its discovery,
BDNF has garnered extensive attention as one of the most extensively studied neurotrophins, owing to its multipotent impacts on various physiological and pathological functions within the nervous system. Recognized for its robust protective actions promoting brain homeostasis, neuronal survival, synaptogenesis, plasticity, and cognitive function
[16][19][20][21][16,19,20,21],
BDNF exhibits activity throughout all stages of development and aging
[22][23][22,23].
BDNF plays a crucial role in initiating compensatory processes that facilitate recovery and/or alleviate chronic adverse effects caused by injury or disease in the nervous system
[19].
Like other neurotrophins, BDNF is initially synthesized as a pre-pro-protein. The pre-protein undergoes rapid cleavage to form pro-BDNF, which then assembles in homodimers
[24][26]. The pro-BDNF can be subsequently cleaved by extracellular proteases at synapses and converted to mature BDNF
[25][27]. BDNF exerts its biological functions via two distinct classes of receptors: the high-affinity tropomyosin receptor kinase B (TrkB) and the low-affinity p75 neurotrophin receptor (p75NTR). Pro-BDNF exhibits a preference for binding to p75NTR, while mature BDNF preferentially binds the TrkB receptor. In general, binding to TrkB receptors allows BDNF to modulate and promote neuronal survival, neuroprotection, and long-term potentiation (LTP)—a form of long-term synaptic plasticity in nociceptive pathways
[26][28]. Conversely, binding to p75NTR receptors may regulate neuronal apoptosis, axonal process pruning, and long-term depression (LTD)
[27][28][29,30]. The contrasting effects of BDNF/TrkB and BDNF/p75NTR signaling form a delicate “yin-yang” system that finely manipulates neuroplasticity and neuronal excitability
[29][31].
The functionality of the central nervous system (CNS) is intricately tied to available
BDNF expression. Predominantly synthesized and expressed in various neuronal cells of the brain, such as sensory neurons and motor neurons
[30][31][32][33,34,35], BDNF is also produced, to a lesser extent, in non-neuronal cells such as glial cells and immune cells
[33][34][36,37]. Available BDNF was found in different regions of the brain, including the neocortex, pyriform cortex, amygdala, hippocampus, claustrum, thalamus, striatum, hypothalamus, and brainstem
[30][35][33,38]. In addition, circulating BDNF derives from both peripheral and cerebral sources
[36][37][38][39,40,41].
3. The Role of BDNF in Patients with Chronic Pain
3.1. The Role of BDNF in Central Sensitization
Chronic pain is known to be associated with CS, a process by which the nociceptive signals of neurons at every level of nociceptive pathways are gradually enhanced
[39][53]. CS is responsible for both hyperalgesia and allodynia. At the cellular level, CS occurs in part as a result of enhanced and more efficient synaptic communication between neurons, which primarily involves the reshaping of neuronal circuits, neuronal hyperexcitability, and a reduction in synaptic inhibition
[40][41][54,55].
Given its essential role throughout the nervous system,
BDNF has been implicated in the induction and maintenance of the CS. For example, the activation of BDNF/TrkB signaling has been linked to increased pain signaling mechanisms
[42][57]. BDNF, upon release from the dorsal root ganglia, engages with TrkB receptors located on primary afferent nerve endings and post-synaptic tracts in the spinal cord. This interaction serves to amplify and potentiate ascending sensory signals, contributing to the perpetuation of CS. As expected, pain signaling mechanisms can be reversed through intrathecal administration of TrkB inhibitors, which attenuates nociceptive response
[43][44][58,59]. It is crucial to recognize that CS involves an activity-dependent increase in the excitability of dorsal horn neurons
[40][54], and
BDNF contributes to this process by promoting a gradual increase in neuronal excitability and synaptic plasticity in the spinal dorsal horn
[45][46][47][60,61,62].
Persistent CS has been described as a maladaptive neuroplasticity process in chronic pain
[48][49][65,66].
BDNF can regulate synaptic plasticity in an activity-dependent manner, contributing to LTP
[50][51][67,68]. LTP involves neuronal adaptation at the presynaptic (e.g., increased ability to produce neurotransmitters) and postsynaptic (e.g., increased ability to bind neurotransmitters to receptors) levels, resulting in enhanced synaptic efficiency and, consequently, an increase in the excitability of neuronal pathways
[52][53][54][69,70,71]. The synapses are a critical link in inter-neuronal connections, and an increase in their number can facilitate the transmission of nociceptive signals between neurons, potentially contributing to CS
[55][72]. However,
BDNF knockout specimens exhibited a decrease in preganglionic synaptic innervation density to sympathetic neurons, suggesting that
BDNF has the ability to increase synaptic density
[22].
Finally, dysfunction in the descending inhibitory nociceptive modulation pathways emerges as a crucial contributor to CS. Recent studies have unveiled reduced intracortical inhibition in different pain populations compared to healthy subjects, with this reduction being associated with more severe pain symptoms
[56][57][58][59][77,78,79,80]. Disinhibition of GABAergic and glycinergic synaptic transmission in nociceptive circuitry is crucial to the generation of chronic pain. Centrally,
BDNF can weaken GABAergic inhibitory synapses by reducing the expression of potassium-chloride cotransporter 2 (KCC2), thus suppressing the intrinsic inhibitory circuits
[46][60][61][61,81,82].
3.2. Neuroinflammation Drives Chronic Pain via Glial-Derived BDNF and CS
While acute inflammation is responsible for triggering acute pain sensations, neuroinflammation is supposed to play an important role in the chronification and persistence of pain
[62][63][84,85]. This neuroinflammation is initiated by the activity-dependent release of glial activators, including neurotransmitters, chemokines, and proteases. This release stems from the central terminals of primary afferent neurons or is prompted by the disruption of the blood–brain barrier. Neuroinflammation is characterized by the activation of glial cells such as microglia and astrocytes, the infiltration of immune cells, vasculature changes, and an increased release of inflammatory and glial mediators like cytokines, chemokines, and BDNF
[64][86]. These glial mediators can significantly regulate both excitatory and inhibitory synaptic transmission, thereby contributing to CS and enhanced chronic pain states.
In the spinal cord and brain, glial cells also produce nerve growth factors and neurotrophins, such as BDNF and basic fibroblast growth factor (bFGF), which can affect neuronal function and may contribute to neurotoxicity in several brain pathologies
[19]. In fact, the expression of neurotrophins is often upregulated in chronic inflammatory diseases due to their involvement in energy homeostasis
[65][88]. For example, microglial activation following peripheral nerve injury upregulates purinergic receptors, especially P2 × 4R, leading to p38-MAPK phosphorylation and subsequent BDNF release
[66][89]. This microglial-derived BDNF has been implicated in facilitating neuropathic pain and morphine hyperalgesia
[67][68][90,91].
A key player in inflammatory activation is the nuclear factor-kappa B (NF-κB), a transcription factor that triggers the expression of pro- and anti-apoptotic genes
[69][99]. Remarkably, the binding of BDNF to the TrkB receptor serves as a trigger for the induction of the NF-κB expression. Furthermore, chronic inflammatory pain has been reported to induce an upregulation of TrkB mRNA and protein expression in the dorsal horn
[70][100]. An additional layer of complexity arises from a p75NTR-mediated effect on NF-κB expression, as evidence suggests that peripheral inflammation induces an upregulation of pro-BDNF and p75NTR in the spinal cord
[71][72][101,102]. With the activation of p75NTR, pro-BDNF can activate several downstream signaling pathways, including extracellular signal-regulated kinase (ERK)1 and ERK2, NF-κB, and c-Jun N-terminal kinase (JNK) pathways, further promoting the neuroinflammatory state
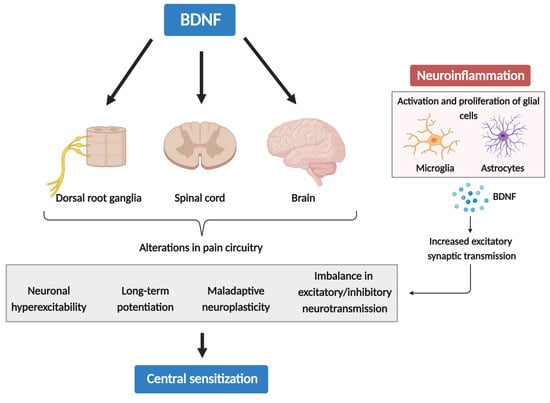
[73][74][75][103,104,105]. These signaling pathways can trigger a series of changes, including neuronal hyperexcitability, LTP, maladaptive neuroplasticity, and an imbalance in excitatory/inhibitory neurotransmission—all of which are intricately involved in the process of CS (
Figure 1). This cycle persists as long as the stressor exists, potentially evolving into a serious chronic pain state. Hence, neuroinflammation may drive chronic pain via CS, which can be induced and maintained by cytokines, chemokines, BDNF, and other glia-produced mediators.
Figure 1. The role of BDNF in central sensitization and neuroinflammation.
3.3. Pro-Nociceptive and Anti-Nociceptive Role of BDNF
Emerging evidence from human studies has revealed higher cerebrospinal fluid [76], plasma [77][78][79], and serum [80][81][82][83] levels of BDNF in patients with chronic pain compared to healthy individuals, which were positively correlated with more severe pain symptoms. For instance, higher serum BDNF levels were associated with lower pressure pain thresholds in patients with fibromyalgia [83]. Notably, a one-month treatment with duloxetine (an antidepressant) not only alleviated pain but also led to reduced serum BDNF levels [84], supporting a pro-nociceptive role of The role of BDNF in central sensitization and neuroinflammation.
3.3. Pro-Nociceptive and Anti-Nociceptive Role of BDNF
Emerging evidence from human studies has revealed higher cerebrospinal fluid [106], plasma [107,108,109], and serum [110,111,112,113] levels of BDNF in patients with chronic pain compared to healthy individuals, which were positively correlated with more severe pain symptoms. For instance, higher serum BDNF levels were associated with lower pressure pain thresholds in patients with fibromyalgia [113]. Notably, a one-month treatment with duloxetine (an antidepressant) not only alleviated pain but also led to reduced serum BDNF levels [114], supporting a pro-nociceptive role of in chronic pain. Recent evidence also supports the pro-nociceptive role of BDNF in arthritis pain [85], with higher plasma BDNF levels observed in patients with knee osteoarthritis compared to healthy controls, positively correlating with self-reported pain levels [79]. BDNF and TrkB were identified in nerve fascicles within synovial tissue from both patients with osteoarthritis and animal models of inflammatory arthritis [86][87]. As expected, experimental injection of peripheral BDNF increased pain behavior [87].
Despite previous studies indicating a strong involvement of
in arthritis pain [115], with higher plasma BDNF levels observed in patients with knee osteoarthritis compared to healthy controls, positively correlating with self-reported pain levels [109]. BDNF and TrkB were identified in nerve fascicles within synovial tissue from both patients with osteoarthritis and animal models of inflammatory arthritis [116,117]. As expected, experimental injection of peripheral BDNF increased pain behavior [117].
Despite previous studies indicating a strong involvement of BDNF in the nociceptive system, its precise role remains uncertain. This uncertainty is further compounded by conflicting findings, with some research indicating a potential anti-inflammatory effect [88][89][90]. For instance, preliminary evidence from animal research suggests that the release of BDNF can alleviate allodynia and hyperalgesia induced by chronic constriction injury [91]. Additionally, in the nociceptive system, its precise role remains uncertain. This uncertainty is further compounded by conflicting findings, with some research indicating a potential anti-inflammatory effect [118,119,120]. For instance, preliminary evidence from animal research suggests that the release of BDNF can alleviate allodynia and hyperalgesia induced by chronic constriction injury [121]. Additionally, BDNF shows anti-inflammatory effects on the animal brain [92], and experimentally induced inflammation, such as the infusion of IL-1 into the hippocampus, and diminishes shows anti-inflammatory effects on the animal brain [122], and experimentally induced inflammation, such as the infusion of IL-1 into the hippocampus, and diminishes BDNF transcription capacity [93]. Several explanations may account for its potential analgesic effect. Firstly, transcription capacity [123]. Several explanations may account for its potential analgesic effect. Firstly, is involved in the regulation of neural circuits, and alterations in neural circuitry may affect inflammatory responses. Emerging evidence suggests that BDNF can inhibit neuroinflammation and regulate cognitive functions [94].
can inhibit neuroinflammation and regulate cognitive functions [124].
3.4. Genetics and BDNF in Chronic Pain
Over the past decade, the identification of altered BDNF levels in individuals with chronic pain has guided many genetic studies, revealing these alterations to be largely genetically determined. Specifically, mutations in the BDNF gene have been found to downregulate its secretion and expression, thereby diminishing its impact on the nervous system [95]. The single-nucleotide polymorphism rs6265 in the gene have been found to downregulate its secretion and expression, thereby diminishing its impact on the nervous system [132]. The single-nucleotide polymorphism rs6265 in the BDNF
gene, located in the 5′-prodomain of immature BDNF protein and often referred to as Val66Met [95][96], has emerged as a key player in shaping pain perception and pain-related symptoms [97][98], and is associated with vulnerability to different chronic pain disorders [99][100][101]. The Val/Val genotype has been linked to a distinct propensity for fibromyalgia symptoms and increased pain catastrophizing [100]. protein and often referred to as Val66Met [132,133], has emerged as a key player in shaping pain perception and pain-related symptoms [134,135], and is associated with vulnerability to different chronic pain disorders [136,137,138]. The Val/Val genotype has been linked to a distinct propensity for fibromyalgia symptoms and increased pain catastrophizing [137].
BDNF, with its influence on crucial neuronal processes, is subject to complex changes in function due to its polymorphism, particularly in modulating neuroplasticity [51]. Evidence suggests that , with its influence on crucial neuronal processes, is subject to complex changes in function due to its polymorphism, particularly in modulating neuroplasticity [68]. Evidence suggests that BDNF polymorphisms can serve as predictors for responses to experimental pain stimulation and non-invasive brain stimulation techniques, contributing to large interindividual variability in stimulation effects [102][103]. polymorphisms can serve as predictors for responses to experimental pain stimulation and non-invasive brain stimulation techniques, contributing to large interindividual variability in stimulation effects [141,142].
3.5. The Epigenetic Regulation of BDNF Expression in Chronic Pain
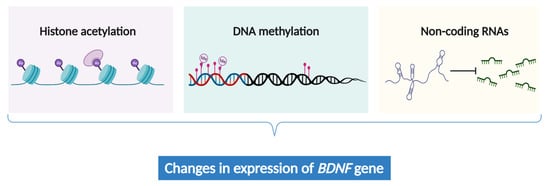
Chronic pain intricately involves abnormal gene expression within the neural cells responsible for processing nociceptive signals in the brain [104][105]. While genetic alterations offer a partial explanation for chronic pain, the emerging field of epigenetics provides a more nuanced and dynamic perspective by unraveling the gene expression patterns associated with chronic pain [106][107]. Recent studies revealed that epigenetic mechanisms, including histone acetylation [108], non-coding RNAs [108][109], and DNA methylation [110][111][112], can influence the expression of Chronic pain intricately involves abnormal gene expression within the neural cells responsible for processing nociceptive signals in the brain [152,153]. While genetic alterations offer a partial explanation for chronic pain, the emerging field of epigenetics provides a more nuanced and dynamic perspective by unraveling the gene expression patterns associated with chronic pain [154,155]. Recent studies revealed that epigenetic mechanisms, including histone acetylation [156], non-coding RNAs [156,157], and DNA methylation [127,158,159], can influence the expression of BDNF
(Figure 2
). These epigenetic modifications may contribute to the pathogenesis and symptomatology of chronic pain.
Figure 2. The epigenetic regulation of
BDNF expression.
4. Clinical and Methodological Implications
4.1. BDNF Treatment for Chronic Pain in a Broader Picture
BDNF
serves as a driving force behind neuroplasticity in the context of chronic pain, positioning it as a potential biomarker and a novel therapeutic target. Although our understanding of BDNF
’s role in pain processing remains limited, emerging evidence suggests its pro-nociceptive involvement in initiating and sustaining CS among individuals with persistent pain. Consequently, exploring the pharmacological and non-pharmacological manipulation of BDNF
opens up crucial avenues for research. Various therapeutic strategies known to influence the release of BDNF have been extensively studied for regulating BDNF levels in patients with chronic pain, including neuromodulation techniques, BDNF
-blocking therapies, and exercise therapy.
Neuromodulation techniques, such as transcranial direct current stimulation (tDCS), emerge as a promising treatment with analgesic properties [113][114][115]. By interfering with ongoing neural activity associated with pain processing and manipulating neuroplasticity and cortical excitability in specific brain regions, tDCS has been reported to improve pain and pain-related symptoms in patients with chronic pain [116][117]. While the exact mechanisms underlying these effects remain unclear, accumulating research suggests that the impact of tDCS may be neuroplasticity-state-dependent [118][119], with alterations in BDNF levels predicting the effects of tDCS on behavioral outcomes [120][121]. In other words, the analgesic effect of tDCS may depend on changes in endogenous Neuromodulation techniques, such as transcranial direct current stimulation (tDCS), emerge as a promising treatment with analgesic properties [167,168,169]. By interfering with ongoing neural activity associated with pain processing and manipulating neuroplasticity and cortical excitability in specific brain regions, tDCS has been reported to improve pain and pain-related symptoms in patients with chronic pain [170,171]. While the exact mechanisms underlying these effects remain unclear, accumulating research suggests that the impact of tDCS may be neuroplasticity-state-dependent [172,173], with alterations in BDNF levels predicting the effects of tDCS on behavioral outcomes [174,175]. In other words, the analgesic effect of tDCS may depend on changes in endogenous levels [176,177], as BDNF is a driving force behind neuroplasticity [49]. is a driving force behind neuroplasticity [66].
Exercise therapy seems to hold the capability to influence BDNF
expression. Recent insights from a systematic review and meta-analysis within pain populations reveal an upregulation of BDNF expression in peripheral blood following diverse physical activities, accompanied by decreasing pain severity [124][125][126]. Similarly promising outcomes have been observed in healthy individuals [127]. However, the duration of exercise can yield varied results, with a single session or acute exercise reportedly increasing BDNF levels, while long-term or regular exercise may reduce them [128][129][130]. expression in peripheral blood following diverse physical activities, accompanied by decreasing pain severity [180,181,182]. Similarly promising outcomes have been observed in healthy individuals [183]. However, the duration of exercise can yield varied results, with a single session or acute exercise reportedly increasing BDNF levels, while long-term or regular exercise may reduce them [184,185,186].
4.2. Using BDNF as an Objective Biomarker
Peripheral blood BDNF has been proposed as a potential biomarker related to disease activity and neuroprogression in various diseases [131][132][133], speculated to mirror alterations in brain expression of Peripheral blood BDNF has been proposed as a potential biomarker related to disease activity and neuroprogression in various diseases [195,196,197], speculated to mirror alterations in brain expression of BDNF. This intricate relationship between brain and blood BDNF levels underscores the potential utility of peripheral measurements as informative markers for CNS dynamics. Given the challenges in directly measuring BDNF levels in the human brain, most clinical studies resort to using plasma or serum samples as proxies [134][135]. . This intricate relationship between brain and blood BDNF levels underscores the potential utility of peripheral measurements as informative markers for CNS dynamics. Given the challenges in directly measuring BDNF levels in the human brain, most clinical studies resort to using plasma or serum samples as proxies [47,198].
BDNF
polymorphisms have emerged as promising pain biomarkers. Specifically, the BDNF Val66Met polymorphism has been detected in diverse chronic pain populations, providing valuable insights into the susceptibility to distinct chronic pain conditions and the considerable interindividual variations in responses to various pain therapies [100][136][137]. Importantly, current literature suggests that Val66Met polymorphism has been detected in diverse chronic pain populations, providing valuable insights into the susceptibility to distinct chronic pain conditions and the considerable interindividual variations in responses to various pain therapies [137,145,200]. Importantly, current literature suggests that BDNF
polymorphisms can be reliably measured in both peripheral blood and buccal swab samples, making them accessible for potential diagnostic applications. Moreover, the development of tests to detect and define chronic pain conditions in the presence of the Val66Met polymorphism is an intriguing prospect. Several techniques, such as genotyping assays or real-time polymerase chain reaction (PCR) methods, could be explored to identify this specific genetic variant in blood samples.
The absence of biomarkers for diagnosing chronic pain remains a significant challenge in clinical practice. Typically, pain severity is assessed through the patient’s subjective report, an approach constrained by difficulties in quantification, reliability, and interparticipant comparability. The integration of objective biomarkers directly linked to the presence and severity of chronic pain would significantly (a) enhance the diagnosis and classification of pain pathophysiology, (b) assist with disease prognostication or predicting therapy responses, and (c) facilitate the development of innovative, mechanism-based treatment approaches, thereby reducing the reliance on long-term opioid use. Overall, BDNF
is one of the most promising biomarkers for chronic pain disorders; however, a definitive clinical validation is still lacking.