1. Introduction
Proteases assume a pivotal enzymatic role, encompassing a broad spectrum of intricate cellular processes such as cellular proliferation, differentiation and the orchestration of programmed cell death
[1][2][1,2]. They recognize specific protein or peptide substrates and cleave amide bonds in a highly regulated manner. Protease activity relies on a multifaceted interplay of factors, including small molecules, cofactors, post-translational modifications and, occasionally, initial cleavage of an inactive zymogen by another protease
[2]. This elaborate regulatory framework serves as a robust mechanism to ensure tight control over enzyme function. Proteases are categorized into six classes based on their proteolytic mechanisms: serine, threonine, cysteine, glutamic, aspartic and metalloproteases
[3]. Among these, serine proteases, which derive their name from the pivotal catalytic serine residue, constitute around a third of the protease population in homo sapiens
[3]. FAM111A, also known as FAM111 trypsin-like peptidase protein, is a serine protease recently explored for its role within human cells
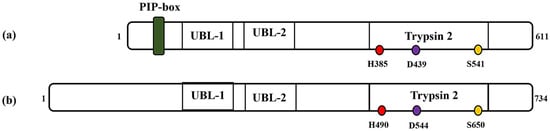
[4]. It is a 611 amino acids protein with a molecular weight of about 70 kDa. The C-terminal region of the human FAM111A protein harbors a serine protease domain reminiscent of the Trypsin 2 enzyme, complete with a conserved catalytic triad composed of His385, Asp439 and Ser541 (
Figure 1). Recently, researchers confirmed FAM111A’s protease activity by conducting in vitro experiments with its recombinant form
[5]. Its auto-catalytic event was elaborated upon in cells overexpressing FAM111A, pinpointing a significant cleavage site between Phe334 and Gly335
[5]. FAM111A encompasses additional structural domains, which include a PCNA (proliferating cell nuclear antigen) interacting peptide (PIP) box
[6] and two ubiquitin-like domains (UBL-1 and UBL-2)
[7]. The PIP box allows FAM111A to interact with DNA replication factors, specifically PCNA, whilst the UBL domains play a role in protein–protein interactions. FAM111A is mainly associated with various functions such as antiviral defense
[8][9][10][8,9,10], DNA replication
[11][12][11,12] and genetic disorders
[13][14][13,14], but its precise role in these diseases is still not fully understood.
Figure 1. Schematic representations of (a) FAM111A and (b) FAM111B protein. PIP: PCNA-interacting peptide box; UBL-1: Ubiquitin-like domain 1; UBL-2: Ubiquitin-like domain 2; Trypsin 2: Trypsin-like peptidase domain. Histidine, aspartic acid and serine amino acid (catalytic triads) are depicted in red, purple and yellow, respectively. The numbers indicate the protein length (1–611 for FAM111A and 1–734, FAM111B) and position of the catalytic amino acid residues (i.e., catalytic triads) on each protein.
FAM111B, a paralog of FAM111A, is a 734 amino acids (85 kDa) protein that exhibits a notable 45% sequence similarity with the FAM111A protein
[6]. Before the recent validation of FAM111B protease activity, bioinformatics studies have indicated the likely existence of a trypsin/cysteine protease-like domain located at the C-terminal region of the FAM111B protein
[4][7][4,7]. Moreover, studies suggest that FAM111B exhibits structural resemblances to FAM111A but notably lacks the PIP box sequence (
Figure 1). Instead, it interacts with PCNA through the replication factor C complex (RFC)
[4][7][4,7]. Furthermore, the FAM111B gene is linked to a reduction in the stability and integrity of the genome, indicating that the gene plays a role in maintaining DNA repair and genome integrity
[15]. Consequently, mutations in this specific gene contribute to advancing cancer and fibrosis
[16].
Apart from the similarities in the structural and catalytic activity of FAM111A and FAM111B, there appears to be an overlap in their cellular function, as indicated in previous studies
[4][6][4,6]. For instance, it was previously shown that mutations in both genes hyperactivated their intrinsic protease activity, which triggered replication and transcription shutdown, disruption of microtubule networks and apoptosis in cells. However, both genes in their wild-type forms and physiological levels are not essential in these cellular processes
[4]. Moreover, co-immunoprecipitation studies carried out in recent studies validated their connectedness
[4][6][4,6]. Nonetheless, the implication of these genes in different genetic disorders diseases suggests that each disease participates in other unique pathways that lead to these diseases and warrants studying these genes individually or collectively in each context or overlapping clinical phenotypes to understand the molecular pathophysiology brought about by their dysregulation and potential implications for therapeutic interventions.
2. The Biological Role of FAM111A and FAM111B in Cellular Processes
2.1. FAM111A at the Replication Fork
Proteomics research suggests that FAM111A is enriched on newly synthesized DNA. FAM111A employs its PIP box to target replication forks by interacting with PCNA
[11][12][11,12]. PCNA acts as a “sliding clamp” that encircles the DNA double helix at replication forks. This encirclement helps DNA polymerase enzymes in place for synthesizing new DNA strands while attached to the DNA template during replication (
Figure 2a). FAM111A also protects replication forks from stalling at protein obstacles caused by nucleoprotein complexes or DNA–protein crosslinks (DPCs) on replicating cells’ DNA
[12] (
Figure 2b). A study conducted by Kojima et al. (2020) revealed that the levels of specific DPCs: topoisomerase 1 cleavage complexes (TOP1ccs) and poly (ADP-ribose) polymerase 1 (PARP1) DNA complexes (i.e., PARP1-DNA), increased in FAM111A knockout (FAM111A KO) cells
[5]. The increase in these DPCs led to the increased sensitivity of FAM111A KO cells to TOP1 and PARP1 inhibitors camptothecin and niraparib, respectively, compared to the wild-type, providing evidence for FAM111A’s important role in genomic stability during replication. More importantly, the same study showed that cells expressing an active site mutant form of FAM111A (i.e., S541A) failed to rescue the replication defects caused by camptothecin and niraparib, suggesting that the protease activity of FAM111A is critical for eliminating these DPCs.
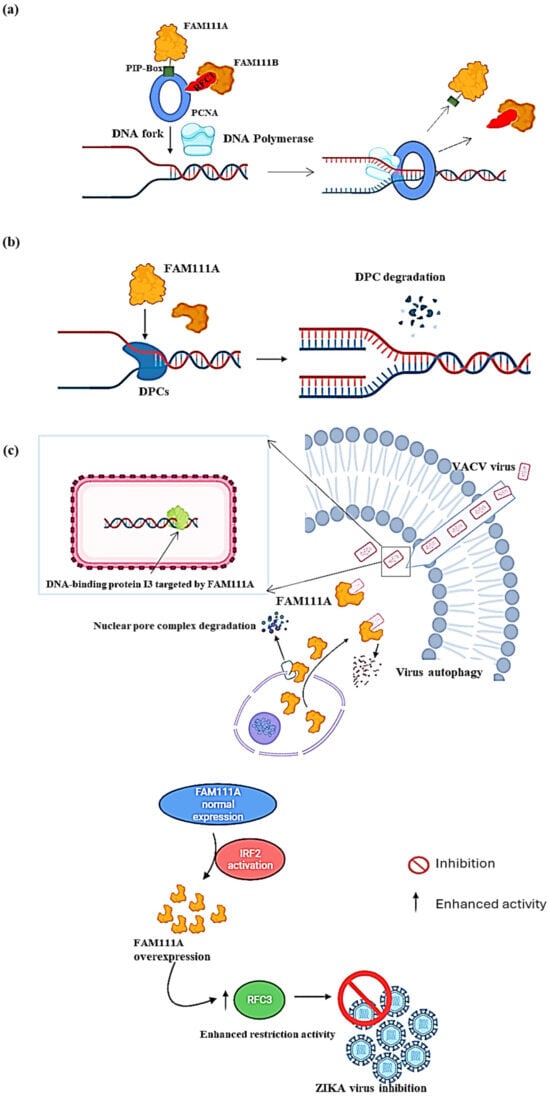
Figure 2. Molecular function of FAM111A. (
a) At the replication fork (PCNA loading). FAM111A helps load PCNA at the replication fork using its PIP box. Once the PCNA is loaded, DNA polymerase can firmly adhere to the DNA strand and progress with DNA replication. (
b) At the replication fork (DPCs degradation). FAM111A targets DNA–protein crosslinks (DPCs) to prevent stalling the replication process or causing DNA breaks. After locating the DPCs, FAM111A uses its proteolytic abilities to degrade them, which allows for the progression of DNA replication. (
c) As a viral replication restriction factor. In the DNA virus VACV (
top panel), FAM111A translocates into the cytoplasm by degrading the nuclear pore complex of the cell by its protease activity and then targets the virus DNA-binding protein I3 for degradation via autophagy. In the RNA virus ZIKA (
bottom panel), FAM111A overexpression is induced by the activation of interferon regulatory factor 2 (IRF2), leading to replication factor C subunit 3 (RFC3) activity enhancement in host cells, thereby increasing host viral restriction activity and ultimately inhibiting ZIKA virus replication. Image created with Biorender (
https://app.biorender.com/illustrations/626a882bceb6a1798ccc52cc, accessed on 3 January 2024).
2.2. FAM111A as a Viral Replication Restriction Factor
FAM111A, a nuclear protein primarily linked to the regulation of host DNA replication, assumes a significant function as an antiviral agent
[8][9][8,9]. Notably, it exhibits antiviral activities against DNA and RNA viruses
[10][17][18][10,17,18], shedding light on its broad-spectrum antiviral potential. In the context of DNA viruses, such as the vaccinia virus (VACV), two studies showed that FAM111A prevented VACV replication by targeting its DNA-binding protein, I3, for autophagic degradation
[9][19][9,19]. This antiviral activity was subsequently shown to be counteracted by the serine protease inhibitor 1 (SPI-1), which is conserved across all orthoviruses
[9][19][9,19], suggesting that FAM111A’s antiviral properties resulted from its proteolytic activity. Moreover, the study by Zhu et al. showed that FAM111A translocated into the cytoplasm upon VACV infection by degrading the nuclear pore complex through its protease activity (
Figure 2c, top panel)
[9]. FAM111A was also shown to exert its influence on the infection of SV40, also known as Simian Virus 40 (SV40), by serving as a factor that restricts the range of hosts. The viral replication process heavily relies on the presence of the SV40 large T antigen (LT), particularly its C-terminal region, which is deemed crucial for lytic infection in specific cell types
[18]. In-depth examination of gene expression and mass spectrometry revealed a distinct interaction between the C-terminal region of LT and FAM111A, and depletion of FAM111A led to heightened viral gene expression and lytic infection of SV40 host range mutants, as well as an escalated replication of adenovirus in cells with restrictive properties
[10]. Similarly, the antiviral activity of FAM111A in RNA viruses was demonstrated in Zika virus (ZIKV)-infected A549 cells
[17]. FAM111A contributed to the inhibition of ZIKV replication through its overexpression, which was induced by the activation of interferon regulatory factor 2 (IRF2), which, in turn, enhanced the host restriction activity of replication factor C subunit 3 (RFC3) to ultimately prevent viral replication, indicating the potential broader applicability of FAM111A-dependent antiviral mechanisms across virus families (
Figure 2c, low panel)
[17].
These findings from this stundedy and similar studies underscore the versatile and critical role of FAM111A as a viral host restriction factor predominantly through its trypsin-like domain and partly its DNA-binding domain, which enables it to bind and interact with viral proteins. Therefore, studying FAM111A’s interactions within a broader range of viruses holds promise for uncovering additional facets of its antiviral function and the potential development of therapeutic strategies targeting these interactions.
2.3. FAM111B, an Integral Player in Regulating DNA Repair or Replication, Cell Cycle and Apoptosis Regulation
FAM111B gene mutations have also been reported to affect genome stability and integrity adversely
[15], contributing to the growing evidence that FAM111B plays a role in maintaining DNA repair and genome integrity. In response to the formation of DPCs or apparent DNA damage, the transcription factor protein p53 activates specific genes that lead to either cellular apoptosis or cell cycle arrest
[20][21][20,21]. Furthermore, the direct or indirect activation of FAM111B by p53 has also been reported
[22]. Moreover, it has been previously shown that the knockout or silencing of FAM111B affects the expression of p53 and p53-related proteins, such as p16, BAG3, BCL-2, CCNB1
[22][23][24][22,23,24], AKT and caspase-1
[23]. Therefore, in addition to FAM111B’s role in DNA repair, its proposed cellular function can be extended to cell cycle regulation.
Initial evidence indicating that FAM111B plays a role in the cell cycle came from a multiomics study that showed a gradual rise in FAM111B RNA levels throughout the G1 phase, ultimately resulting in the noticeable accumulation of the FAM111B protein during the S phase
[21]. These findings were validated in a lung adenocarcinoma (LUAD) cell line where FAM111B had been deleted
[25]. Notably, there was a marked decrease in the number of cells progressing through the S and G2/M phases of the cell cycle, and a higher proportion of cells remained in the G0/G1 phases. FAM111B was also shown to influence the degradation of p16 (CDKN2A), a protein responsible for delaying the G1/S transition by inhibiting the formation of the CDK 4/6—Cyclin D complex
[24][26][24,26]. This complex is responsible for phosphorylating the retinoblastoma protein (pRb1), allowing for its dissociation from E2F and promoting cell cycle progression. If the DNA repair process is effective, FAM111B could initiate the resumption of the cell cycle by decreasing the levels of p16 (
Figure 3a)
[16][26][16,26].
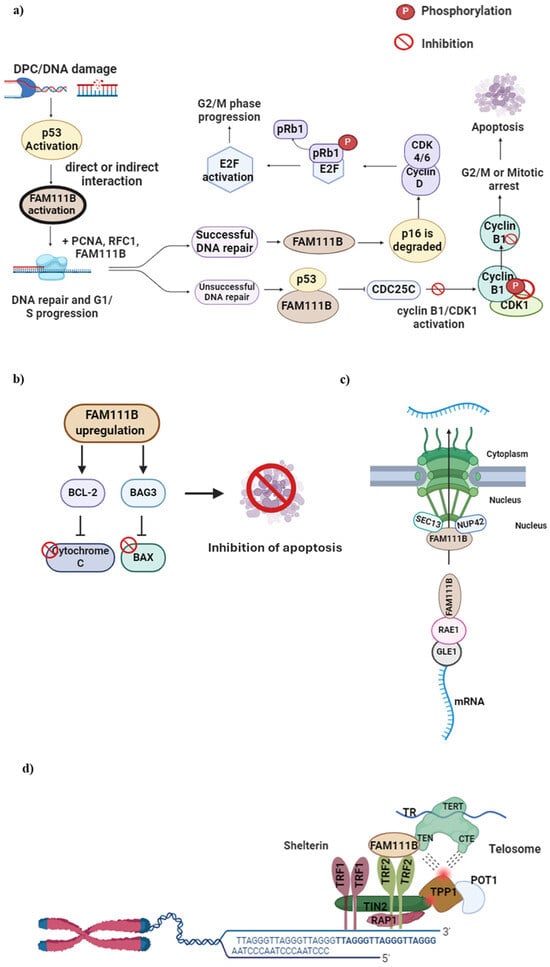
Figure 3. FAM111B functions in DNA repair, cell cycle, apoptosis, nuclear transport and telomere maintenance. (
a) DPC formation or DNA damage in the G1 to S phase activates the p53 protein, which, in turn, activates FAM111B. Upon successful DNA repair, FAM111B degrades p16, leading to CDK 4/6—Cyclin D complex formation. This complex phosphorylates pRb1 and dissociates it from E2F, which then becomes activated, allowing cell cycle progression to G2/M. When DNA repair is unsuccessful, FAM111B and p53 inhibit C Cyclin B, resulting in delayed cell cycle progression into the G2/M phase. (
b) FAM111B upregulation activates BCl-2 and BAG3, inhibiting pro-apoptotic Cytochrome C and BAX proteins, respectively, preventing cellular apoptosis and cell survival, suggesting FAM111B is involved in apoptosis. (
c) FAM111B may function as a nuclear export protein through interactions with nucleoporins SEC13 and NUP42 and mRNA export via binding to the RNA export proteins RAE1 and GLE1 RNA. (
d) FAM111B functions in telomerase maintenance by recruiting TRF2, a shelterin protein complex component vital for healthy telomeres. Image created with Biorender (
https://app.biorender.com/illustrations/626a882bceb6a1798ccc52cc, accessed on 7 January 2024).
Aligning with the previously proposed and experimentally determined function of FAM111B, a recent study showed that the knockout of FAM111B caused an increase in p16, E2F and pRb1 expression levels
[22] and concluded that FAM111B further regulates the cell cycle by controlling the activity of cyclin B1 and CDC25C. The protein cyclin B1 regulates the cell cycle passage from the G2 to the M phase, which is necessary for healthy cell division. Cyclin B1 binds to and activates CDK1, which sets off a series of processes that lead to mitosis. Activated p53 and, possibly, FAM111B inhibit cyclin B1, thereby delaying the G2/M transition and allowing more time for DNA repair
[16] (
Figure 3).
The function of the FAM111B protein in the S phase of the cell cycle was elucidated by discovering its interaction with DNA-binding proteins like replication factor C subunit 1 (RFC1) and proliferating cell nuclear antigen (PCNA)
[4]. RFC is a complex comprising five subunits vital in loading PCNA onto DNA, a critical step in DNA replication
[27]. The disruption of PCNA and RFC was reported in cells overexpressing FAM111B or cells expressing mutant FAM111B derived from patients
[4], supporting its role in DNA replication.
FAM111B was suggested to influence the apoptotic pathway by upregulating the expression of anti-apoptotic proteins BCL2 and BAG3 (Bcl-2-associated athanogene3)
[22]. BCL2 inhibits the release of cytochrome c into the cytoplasm whilst BAG3 simultaneously interacts with and inhibits the pro-apoptotic protein BAX; in this way, programmed cell death is suppressed, resulting in cell survival
[28][29][28,29]. Furthermore, Sun et al. (2019) showed that FAM111B may bind directly with BAG3 by demonstrating that the expression of FAM111B was decreased with a decreased BAG3 and BCL2 expression
[22]. All these findings provide strong evidence for FAM111B in DNA damage repair and replication, cell division and apoptosis and warrant further studies in this context.
2.4. The Involvement of FAM111B in Nuclear Transport and Telomere Length Maintenance
Studies have reported on the mainly nuclear localization of FAM111B
[4][30][4,30] and thus proposed nucleo-specific roles as previously described
[16]. A recent study on the cellular function of FAM111B showed it interacted with specific components of the nuclear pore complexes, precisely two nucleoporins, SEC13 and NUP42
[31]. This study indicated that FAM111B is recruited to the nuclear periphery through this interaction to perform its function
[31]. Furthermore, FAM111B’s association with these nucleoporins suggests its involvement in nucleo-cytoplasmic trafficking, specifically in mRNA exports. However, the study reported no significant changes in the global mRNA transport in wild-type and FAM111B KO cells
[31]. This study, however, showed by mass spectroscopy that FAM111B interacts with the ribonucleic acid export 1 (RAE1) and GLE1 RNA export mediator (GLE1) proteins, which are components of the RNA export machinery. However, the exact mechanisms remain unclear and warrant further studies to validate this role.
Kliszczak et al. (2023) also showed that FAM111B is essential in maintaining normal telomere length
[31]. In FAM111B KO cells, telomeres were reported to be shorter due to the lower recruitment of the telomere repeat binding factor 2 (TRF2) component of shelterin (or telosome, a protein that protects telomeres and regulates telomerase activity)
[32]. The loss of TRF2 or shelterin has been associated with genome instability and the activation of DNA damage response and repair mechanisms in cells, including end-to-end fusion
[33], non-homology or homologous directed repair
[34][35][34,35], p53 activation and ATM-mediated apoptosis
[36]. In other words, the loss of FAM111B or disease-associated mutation results in the loss of TRF2, leading to the critical shortening of telomeres and genome instability, further providing evidence for FAM111B in this vein.
2.5. The Overlap in the Cellular Functions of FAM111A and FAM111B
The cellular functions of FAM111A and FAM111B exhibit notable overlap, particularly in their roles related to DNA replication, transcription regulation, microtubule organization and apoptotic pathways
[4]. FAM111A dominant missense mutations that hyperactivate its intrinsic protease activity have been shown to disrupt DNA replication, transcription and apoptosis induction
[4]. Similarly, FAM111B heterozygous missense mutations near its protease domain have been demonstrated to elevate its protease activity and subsequent impairment of DNA synthesis, transcriptional processes, microtubule integrity and apoptotic cell death
[4][30][4,30].
Moreover, FAM111A and FAM111B proteins have been shown to interact with cellular replication and transcription components like RFC subunits and RPB1, suggesting potential functional synergy
[4]. Furthermore, a FAM111B-enriched interactome analysis in another study identified FAM111B as a primary interactor of FAM111A
[6], supporting their collaborative roles, possibly by forming a joint complex to regulate various cellular processes. Therefore, despite the differences in the clinical manifestations of disorders associated with FAM111A and FAM111B mutations, their pathological mechanisms converge on the dysregulation of protease activity, highlighting a shared molecular basis underlying their disease-promoting potential.