Neurodegenerative disorders like Alzheimer’s disease (AD), Parkinson’s disease (PD), frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) affect millions of people worldwide, and as the average human lifespan increases, similarly grows the number of patients. Cognitive and motoric decline has been explained by the very apparent deterioration of neurons in various regions of the brain and spinal cord. However, more studies show that disease progression is greatly influenced by the vast population of glial cells. Astrocytes are traditionally considered star-shaped cells on which neurons rely heavily for their optimal homeostasis and survival. Increasing amounts of evidence depict how astrocytes lose their supportive functions while simultaneously gaining toxic properties during neurodegeneration.
- astrocyte
- neurodegenerative diseases
- communication
- synapse
- mitochondrial
- metabolism
- protein aggregates
- autophagy
- glial border formation
1. Introduction
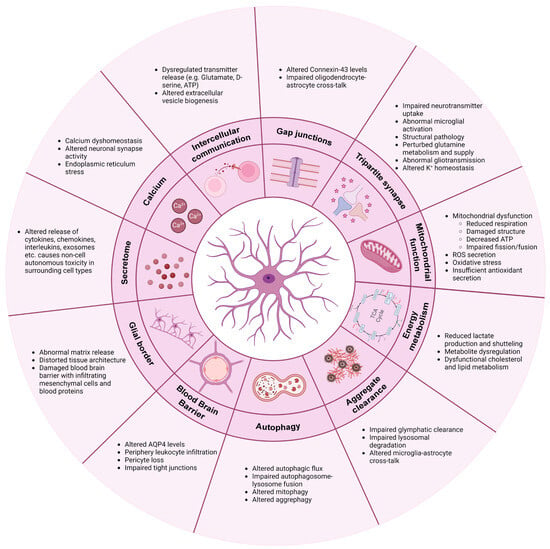
2. Communication
Unlike neurons, astrocytes are non-excitable and therefore have developed other means of communication. Besides cytokine, chemokine and interleukin releases, astrocytes use calcium waves, transmitter release and gap junction couplings as a means of communication to maintain network homeostasis [49][20].2.1. Calcium
Astrocytic calcium transients are highly dynamic. The “waves” are projected intra- and intercellularly through calcium-permeable ion channels and receptors, including various neurotransmitter receptors [5][21]. Upon neurotransmitter binding, a receptor-specific intracellular calcium signal is evoked, which allows astrocytes to distinguish between, e.g., glutamatergic and cholinergic synaptic activity [95,96][22][23]. Individual astrocytic subpopulations display unique calcium waves, which further contributes to their heterogeneity and complexity [97][24]. In multiple neurodegenerative diseases including AD [42,98–102], ALS [103,104], Huntington's disease (HD) [105–107] and PD [29,108], calcium dyshomeostasis is observed. The dysregulated calcium response is often detected early in disease progression and might therefore contribute to the pathologically altered neuronal synapse activity [102,105,107,109–111]. For example, oligomeric forms of Aβ peptide were shown to drive astrocytic calcium hyperactivity early in AD disease progression, which consequently triggered glutamatergic hyperactivity in adjacent neurons [109,110]. Calcium is primarily stored in the endoplasmic reticulum (ER) within astrocytes but can also be imported into the cells through AMPA and N-methyl-D-aspartate (NMDA) receptors or through voltage-gated channels [49]. In ALS and PD, abnormal intracellular calcium dynamics with excess ER accumulation and storage release is observed, ultimately contributing to ER stress [29,103,104]. Both genetic PD mutations and drug-induced depletion of dopamine transmission mimicking PD have shown to increase astrocytic calcium excitability [29,108]. In the R6/2 transgenic mouse model of HD, spontaneous calcium signals and storage capacity are reduced, while evoked calcium responses are increased [105].2.2. Intercellular Communication
In addition to receiving neurotransmitter information, calcium transients can release transmitters such as glutamate, GABA, D-serine and ATP from astrocytic processes, which contribute to the regulation of neuronal synapse excitability and plasticity [49,95][20][22]. In pathological conditions, the release of these transmitters from both neurons and astrocytes is dysregulated, and studies show that excess release causes a continuous self-activation, leading to cytotoxicity [42,99,112][25][26][27]. This phenomenon is observed in several models of PD and AD and is triggered by excessive astrocytic calcium-dependent release of glutamate and D-serine [110,111,113]. Similarly, astrocytic release of ATP can bind purinergic P2Y receptors on microglia, thereby modulating their phagocytic functions and release of cytokines further driving inflammation [114,115]. Astrocytic self-activation is also enhanced by the release of astrocytic TNF-α and prostaglandin, which, through calcium signaling, can promote additional “gliotransmitter” release [112]. The astrocyte–neuron communication can further be impaired through other means such as extracellular vesicle (EV) production. In LRKK2-mutant astrocytes, the EV biogenesis is altered, causing an accumulation of PD-related proteins within multivesicular bodies [116]. These astrocyte-secreted EVs are internalized by dopaminergic neurons and are linked to an astrocytic failure of providing neurotrophic support [116]. Furthermore, C3-containing exosomes are released by AD astrocytes, consequently driving an inflammatory response and being linked to reduced neurite outgrowth [55,117].2.3. Gap Junctions
Astrocytes not only communicate via neurotransmitters but also through gap junction couplings. Connexin-43 (Cx43) is the predominant connexin protein, which constitutes hemichannels and gap junctions in astrocytes [118,119,120][28][29][30]. Through these, diffusion of ions, metabolites, miRNAs and second messengers is facilitated, in addition to the important mitigation of calcium waves [121][31]. Therefore, gap junctions are key components in astrocyte networks, contributing to synapse activity modulation and homeostatic buffering. In ALS [69,122,123], AD [42,124] and PD [108], Cx43 is abnormally elevated, which causes increased gap junction coupling and hemichannel activity, resulting in calcium hyperactivity, neuronal excitability and cell death. Astrocytes also form collaborative glial networks with oligodendrocytes through their Cx43/Cx47 gap junction connections, which are important for their coordinated cross-talk [125]. In MS, oligodendrocytes and astrocytes lose their communication through decreased Cx43/Cx47 gap junction expression, consequently promoting demyelination and inflammation [126–128]. Lack of Cx43/Cx47 gap junction couplings is likewise found in AD [129].3. Tripartite Synapse: Neurotransmitter Regulation and Synapse Function
Astrocytes are highly involved in neuronal synapse plasticity and function through their perisynaptic process ensheathment of neuronal synapses [130][32]. This tripartite synapse collaboration between pre- and postsynaptic neurons and astrocytes ensures optimal neuronal firing by continued astrocytic removal and recycle of excess neurotransmitters from the intersynaptic space [131,132][33][34]. In neurodegeneration, this function is impaired. Due to the downregulation of key astrocyte receptors, excitatory amino acid transporters 1 and 2 (EAAT1 and EAAT2), astrocytes fail to properly manage the uptake of the neurotransmitter glutamate. Glutamate is the main excitatory neurotransmitter in the brain, and a large part of its uptake from the synaptic cleft appears through EAAT1/2 receptors on astrocytes [133,134,135][35][36][37]. Persistent glutamate exposure is believed to cause excessive neuronal firing and abnormal neuronal calcium influx, which ultimately results in severe neuronal excitotoxicity [131,134,136,137][33][36][38][39]. Lack of EAAT1/2 receptors and consequent glutamate excitotoxicity is a common phenomenon in AD [138[40][41][42][43][44],139,140,141,142], PD [143[45][46],144], ALS [48,83,145,146][47][48][49][50] and HD [27,28,105][51][52][53]. Additionally, downregulation of astrocytic glutamate receptors drives abnormal microglial pruning and phagocytosis of hippocampal glutamatergic synapses in AD [147][54]. In HD, structural pathology, mediated by lack of astrocytic engagement of neuronal synapses, enables the hyper-excitability [148][55]. AD astrocytes likewise have perturbed glutamine metabolism and supply, which affects the GABA synthesis [149,150,151,152][56][57][58][59]. GABA is the main inhibitory neurotransmitter in the central nervous system, and lack thereof contributes to the excitatory imbalance [5][21]. In addition to neurotransmitter uptake, astrocytic release of various transmitters mentioned above is additionally important for synapse activity regulation. Abnormal gliotransmission has thus been shown to affect the synaptic transmission in PD and AD, consequently contributing to synapse loss and excitotoxicity [110,113,137,153,154,155][39][60][61][62][63][64]. Similarly, astrocytic release of C3 can bind to neuronal C3 receptors and hamper their synaptic density and dendritic morphology [156][65]. Astrocytes control the ion homeostasis through multiple ion channels, which is crucial for maintaining synapse functionality [157][66]. Potassium (K+) buffering is a key mechanism affected in neurodegeneration. Under physiological conditions, astrocytes regulate K+ levels in the extracellular space through clearance via K+ channels such as the main astrocytic Kir4.1 sub-type [49][20]. Through these mechanisms, astrocytes can modulate neuronal depolarization and thereby excitability [158][67]. In HD [26][68] and ALS [159][69], astrocytic Kir channels are downregulated, which hampers the K+ buffering and clearing, consequently causing increased extracellular K+ levels, overall leading to neuronal excitotoxicity. Besides through Kir.4.1 channels, K+ is released through EAAT2 and as mentioned previously, the astrocytic release and uptake of transmitters is modulated by calcium transients. This interconnected relationship between ion homeostasis, calcium dynamics and gliotransmission is therefore crucial for optimal neuronal function, and any dysregulation of one mechanism might consequently disrupt the others.4. Mitochondrial Function
Mitochondrial dysfunction, reactive oxygen species (ROS) secretion and oxidative stress are common findings in neurodegeneration. In AD, mitochondrial dysfunctions are predicted as an early astrocytic phenotype consequently driving astrocyte reactivity [23][70], and in PD, maintenance of mitochondrial DNA is correlated with reduced mitochondrial respiration [29][71]. Astrocytic accumulations of α-synuclein in PD have been linked to damage within the mitochondrial structure, consequently lowering the total ATP level as well as causing disruption of the fission and fusion dynamics [160,161][72][73]. In ALS, mitochondria display decreased membrane potential and a compromised oxygen consumption, possibly correlated with a lower secretion of antioxidants [85,162][74][75]. Astrocytes express and release antioxidants as a part of their function in regulating the redox balance through removal of ROS in order to prevent oxidative damage of neurons [163][76]. Astrocytic nuclear factor erythroid 2-related factor 2 (Nrf2) transcription factor is a key regulator of antioxidant, detoxification and proteostasis pathways and might therefore be an important mediator in neurodegeneration [163][76]. In amyloid and tau pathology models of AD, pre-emptive activation of astrocytic Nrf2 was shown to be neuroprotective by attenuating the aggregation burden and slowing disease progression [33][77]. Similar observations were seen by overexpressing Nrf2 in astrocytes in SOD1-ALS mice [164][78] and in α-synuclein-mutant mice in PD [165][79], which advocates for a common therapeutic mechanism in neurodegeneration. Interestingly, Nrf2 target gene levels are in fact increased in AD but might appear too limited or too late in the disease progression to make a considerable neuroprotective difference [33,166][77][80].5. Energy Metabolism
Neurons rely on astrocytes for nutritional support to meet their optimal energy consumption. Through key mechanisms such as the lactate shuttle, astrocytes convert glucose or glycogen to lactate via aerobic glycolysis and provide the lactate to adjacent neurons via monocarboxylate transporters [167][81]. In the neurons, the lactate is incorporated into the oxidative cycle for ATP production [167][81]. Lactate is also shuttled directly to neurons from the bloodstream via the astrocytes’ perisynaptic process engagement with the vasculature [168][82]. In ALS, astrocytic intra- and extracellular levels of lactate are decreased, possibly due to diminished lactate production and/or transport [169,170][83][84]. Similarly, AD astrocytes have decreased glycolysis and secretion of lactate to the environment [101][85]. Lack of lactate production causes metabolic dysfunction and failure of adequate neuronal energy supply. Additionally, lactate production is coupled to glutamate transporter activity, and their shared impairment might therefore further hamper the energy metabolism in neurodegenerative disorders [12][86]. A widespread dysregulation of metabolites is generally observed in neurodegenerative diseases. Astrocyte metabolic proteomics is increasingly enriched during the commencement and disease progression of AD, with markers expressing both neuroprotective and neurotoxic phenotypes [24][87]. More specifically, AD astrocytes display augmented glycolytic flux and reduced glycogen storage [142][44]. In PD, astrocytes have altered polyamine and phospholipid levels [29][71], and in ALS, astrocytic metabolic dysfunction with compromised adenosine, fructose and glycogen metabolism is observed [171,172][88][89]. Finally, astrocytes are the main synthesizers and suppliers of apolipoprotein E (ApoE), which ensures sufficient transport of cholesterol to neurons [49][20]. Cholesterol is an essential lipid in cell membranes and presynaptic vesicle formation and is therefore crucial for the integrity and function of synapses. One of the main risk factors of late-onset AD is the human isoform APOEε4 variant [173,174,175,176][90][91][92][93]. Carriers are shown to have pathologically altered glucose metabolism, lactate production, cholesterol homeostasis and calcium dynamics, resulting in AD [98,177,178,179,180][94][95][96][97][98]. Dysfunctional cholesterol and lipid metabolism are also found in HD [35,181,182][99][100][101] and PD [183,184][102][103] and appear to be a general mechanism of reactive astrocytes, potentially influencing many neurodegenerative diseases [185][104].6. Clearance of Protein Aggregates
Astrocytes have been shown to contribute to the insufficient clearance of protein aggregates in neurodegeneration due to failure of the glymphatic system. Under physiological conditions, astrocytes regulate water flux as well as removal of metabolic waste products from the brain interstitium to and from the perivascular space through their Aquaporin-4 (AQP4) water channels [5][21]. Additionally, through the synergistic collaboration between AQP4 and Kir4.1 channels, osmohomeostasis is maintained in the brain [186,187,188][105][106][107]. During disease progressions, astrocytes remove Aβ peptides and mutant huntingtin through the glymphatic system [189,190][108][109]. However, due to the downregulations of Kir4.1 and AQP4, this clearing mechanism is impaired, consequently accelerating the aggregation burden [26,191,192][68][110][111]. Additionally, astrocytic ApoE is normally involved in Aβ clearance, but in APOEε4 carriers, this function appears to be compromised, which likewise contributes to the enhanced Aβ aggregation [179,193][97][112]. Secondly, astrocytes contribute to clearance of proteins through their intracellular lysosomal pathways. This mechanism is affected in PD, where both α-synuclein oligomers as well as mutations in the LRRK2 gene interfere with the astrocytic clearance of the aggregation burden [160,194][72][113]. Neuronal α-synuclein is taken up by astrocytes from the extracellular space via endocytosis as well as transmitted directly from neurons [93,195][18][114]. Similarly, aggregates are transferred between astrocytes via nanotubules [161][73]. This increased inclusion burden aided by overload/stress-induced insufficient lysosomal degradation further triggers cellular toxicity and reactivity [93,161][18][73]. More specifically, α-synuclein is shown to alter the lysosomal morphology, distribution and function by alkalinization and decrease activity of lysosomal proteases in neuronal cells and idiopathic PD brains [196[115][116],197], which correlates with similar observations of disrupted lysosomal proteolysis recently observed in astrocytes in an early-onset PD model [198][117]. Astrocytic engulfment of monomeric tau from the extracellular environment has been observed in tauopathies [199][118], and the endo-lysosomal dysfunction in astrocytes is likewise predicted as an early disease phenotype in AD [23,200][70][119]. Finally, microglial and astrocytic cross-communication is important for the optimal clearance of Aβ and α-synuclein aggregates [201][120]. Intercellular miscommunication or toxic reactivity might therefore hamper this essential function, thereby accelerating disease progressions.7. Autophagy
Autophagy is a vital cellular process, which facilitates the degradation and recycling of intracellular components such as damaged organelles and protein aggregates [202][121]. Several conditions like starvation, stress and pharmacological treatment can modulate autophagy to facilitate a faster release of important nutrients in the reestablishment of cellular homeostasis [203][122]. Autophagy is a multi-step process, where formation of large double-membrane vesicles termed autophagosomes bind and enclose cargo destined for degradation through ubiquitin-P62-LC3 protein interactions [202][121]. The autophagosomes fuse with late endosomes and finally with lysosomes, and the lysosomal enzymes degrade the vesicular content for reuse [204][123]. Mutations in LRRK2 are a common cause of genetic PD [205][124]. LRRK2 contributes, among many functions, to the phosphorylation of various proteins from the Rab family as well as P62, thereby filling a prominent role in autophagy initiation and vesicle transport [206][125]. Consequently, LRRK2-mutant astrocytes display impaired autophagy, possibly linked to progressive accumulation of α-synuclein as mentioned above [207][126]. Additionally, α-synuclein oligomers have been shown to interfere with the autophagosome–lysosome fusion, consequently halting the autophagic flux in astrocytes [161][73]. This degradation impairment results in insufficient turnover of damaged mitochondria [161][73]. Moreover, in ALS/FTD-relevant C9ORF72-mutant models, accumulation of P62 is present in astrocytes [208][127], and we previously showed how human induced pluripotent stem cell-derived FTD astrocytes displayed insufficient autophagy, consequently perturbing the mitochondrial turnover [60][3]. This lack of mitophagy resulted in augmented mitochondrial fusion, impaired mitochondrial respiration and glycolysis, increased ROS and stress granules formation, which overall triggered astrocyte reactivity and cytokine secretion [60][3].8. Blood–Brain Barrier (BBB)
Through close interaction with capillary endothelial cells and pericytes, astrocytes function as efficient gatekeepers of the central nervous system through regulation of the integrity and permeability of the BBB [6][128]. Additionally, neurons rely on astrocytes for shuttling of nutrients and metabolic substrates from the blood stream through the BBB [6][128], and with calcium-dependent release of vasodilators or vasoconstrictors, astrocytes control the blood flow depending on the neuronal energy demand [209][129]. As mentioned previously, AQP4 levels are changed in various neurodegenerative diseases, which also contribute to the compromised BBB [13,210][130][131]. Therefore, cerebrovascular deterioration in AD could be correlated with astrocytic calcium hyperactivity [42][25]. In ALS, astrocytes contribute to BBB disruption and pericyte loss, which allow periphery leukocyte infiltration and a disturbance of the homeostatic environment [43,211,212,213,214][132][133][134][135][136]. In AD, APOEε4 carriers display pericyte degeneration due to insufficient astrocyte suppression of proinflammatory pathways, consequently damaging the BBB [215,216][137][138]. Additionally, the barrier function of BBB tight junctions is impaired when astrocytes carry the APOEε4 isoform [217][139], and astrocytes modulate leukocyte infiltration due to Aβ exposure [218][140]. In HD, astrocytes trigger endothelial cell proliferation and pericyte damage, which disrupts the vascular function [219][141]. Finally, in PD, reactive astrocytes fail to support vessel formation and barrier integrity [220][142].9. Glial Border Formation
Astrocytes are crucial for the integrity of the stroma in the central nervous system and provide important structural support [5][21]. Through their secretion of molecules such as proteoglycans, astrocytes contribute to the extracellular matrix (ECM) structure surrounding synapses and within the synaptic cleft. This is important for capturing nutrients and growth factors as well as acting as a diffusion barrier for neurotransmitter concentration buffering [49][20]. During neurodegeneration, subpopulations of astrocytes initiate a border formation, which primarily functions to contain the injury. However, emerging evidence point towards a rising role of abnormal scarring in these disorders [221][143]. Here, ECM molecules promote a pro-inflammatory signal mediated by microglia and infiltrating macrophages, which further drives the astrocytic reactivity and matrix release [221][143]. Conversely, glial border formation is shown to be beneficial in its formation of “glial bridges”, along which axonal regrowth is permitted if appropriately guided by growth factors [222][144]. Lack of astrocytic neuro-supportive functions likely influences this mechanism in neurodegenerative diseases [34][145]. Chondroitin sulfate proteoglycans (CSPGs) are central components of glial border formation and mediated by reactive astrocytes in MS lesions [223][146]. Although this physical barrier aids in the containment of the lesion, it also prevents axonal outgrowth and remyelination [223][146]. As a consequence, excessive glial border formation by astrocytes might distort the tissue architecture, thereby prolonging the disease course of MS. Similarly, in ALS, CSPG accumulation is present at the site of motor neuron degeneration, and excessive and abnormal CSPG receptors are also found on the surface of reactive astrocytes, further contributing to their self-activation and the overall impairment of the homeostatic environment [224,225][147][148]. Additionally, chronic release of TGF-β promotes excessive fibrosis in ALS [226][149] and acts as an upstream regulator of CSPG secretion [227,228][150][151]. In AD, astrocytic proteoglycans interact with Aβ plaques, consequently promoting aggregation and inhibiting clearance [229][152]. Finally, the STAT3 pathway in astrocytes is an important modulator of glial border formation after spinal cord injury [230][153]. Abnormal expression of the STAT3 pathway in multiple neurodegenerative disease could likely contribute to border formation imbalances. Similarly, the damage to the BBB generally observed in neurodegeneration permits the infiltration of mesenchymal cells and blood proteins, which mediates excessive scarring [221][143].References
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487.
- Hartmann, K.; Sepulveda-Falla, D.; Rose, I.V.L.; Madore, C.; Muth, C.; Matschke, J.; Butovsky, O.; Liddelow, S.; Glatzel, M.; Krasemann, S. Complement 3+-Astrocytes Are Highly Abundant in Prion Diseases, but Their Abolishment Led to an Accelerated Disease Course and Early Dysregulation of Microglia. Acta Neuropathol. Commun. 2020, 7, 83.
- Chandrasekaran, A.; Stoklund Dittlau, K.; Corsi, G.I.; Haukedal, H.; Doncheva, N.T.; Ramakrishna, S.; Ambardar, S.; Salcedo, C.; Schmidt, S.I.; Zhang, Y.; et al. Astrocytic Reactivity Triggered by Defective Autophagy and Metabolic Failure Causes Neurotoxicity in Frontotemporal Dementia Type 3. Stem Cell Rep. 2021, 16, 1–16.
- Guttenplan, K.A.; Weigel, M.K.; Adler, D.I.; Couthouis, J.; Liddelow, S.A.; Gitler, A.D.; Barres, B.A. Knockout of Reactive Astrocyte Activating Factors Slows Disease Progression in an ALS Mouse Model. Nat. Commun. 2020, 11, 3753.
- Hou, B.; Zhang, Y.; Liang, P.; He, Y.; Peng, B.; Liu, W.; Han, S.; Yin, J.; He, X. Inhibition of the NLRP3-Inflammasome Prevents Cognitive Deficits in Experimental Autoimmune Encephalomyelitis Mice via the Alteration of Astrocyte Phenotype. Cell Death Dis. 2020, 11, 377.
- Abjean, L.; Haim, L.B.; Riquelme-Perez, M.; Gipchtein, P.; Derbois, C.; Palomares, M.A.; Petit, F.; Hérard, A.S.; Gaillard, M.C.; Guillermier, M.; et al. Reactive Astrocytes Promote Proteostasis in Huntington’s Disease through the JAK2-STAT3 Pathway. Brain 2023, 146, 149–166.
- Leng, K.; Rose, I.V.L.; Kim, H.; Xia, W.; Romero-Fernandez, W.; Rooney, B.; Koontz, M.; Li, E.; Ao, Y.; Wang, S.; et al. CRISPRi Screens in Human IPSC-Derived Astrocytes Elucidate Regulators of Distinct Inflammatory Reactive States. Nat. Neurosci. 2022, 25, 1528–1542.
- Marchetto, M.C.N.; Muotri, A.R.; Mu, Y.; Smith, A.M.; Cezar, G.G.; Gage, F.H. Non-Cell-Autonomous Effect of Human SOD1G37R Astrocytes on Motor Neurons Derived from Human Embryonic Stem Cells. Cell Stem Cell 2008, 3, 649–657.
- Gomes, C.; Sequeira, C.; Likhite, S.; Dennys, C.N.; Kolb, S.J.; Shaw, P.J.; Vaz, A.R.; Kaspar, B.K.; Meyer, K.; Brites, D. Neurotoxic Astrocytes Directly Converted from Sporadic and Familial ALS Patient Fibroblasts Reveal Signature Diversities and MiR-146a Theragnostic Potential in Specific Subtypes. Cells 2022, 11, 1186.
- Di Giorgio, F.P.; Boulting, G.L.; Bobrowicz, S.; Eggan, K.C. Human Embryonic Stem Cell-Derived Motor Neurons Are Sensitive to the Toxic Effect of Glial Cells Carrying an ALS-Causing Mutation. Cell Stem Cell 2008, 3, 637–648.
- Stoklund Dittlau, K.; Terrie, L.; Baatsen, P.; Kerstens, A.; De Swert, L.; Janky, R.; Corthout, N.; Masrori, P.; Van Damme, P.; Hyttel, P.; et al. FUS-ALS HiPSC-Derived Astrocytes Impair Human Motor Units through Both Gain-of-Toxicity and Loss-of-Support Mechanisms. Mol. Neurodegener. 2023, 18, 5.
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from Familial and Sporadic ALS Patients Are Toxic to Motor Neurons. Nat. Biotechnol. 2011, 29, 824–828.
- Taha, D.M.; Clarke, B.E.; Hall, C.E.; Tyzack, G.E.; Ziff, O.J.; Greensmith, L.; Kalmar, B.; Ahmed, M.; Alam, A.; Thelin, E.P.; et al. Astrocytes Display Cell Autonomous and Diverse Early Reactive States in Familial Amyotrophic Lateral Sclerosis. Brain 2022, 145, 481–489.
- Miller, S.J.; Glatzer, J.C.; Hsieh, Y.C.; Rothstein, J.D. Cortical Astroglia Undergo Transcriptomic Dysregulation in the G93A SOD1 ALS Mouse Model. J. Neurogenet. 2018, 32, 322–335.
- Ziff, O.J.; Clarke, B.E.; Taha, D.M.; Crerar, H.; Luscombe, N.M.; Patani, R. Meta-Analysis of Human and Mouse ALS Astrocytes Reveals Multi-Omic Signatures of Inflammatory Reactive States. Genome Res. 2022, 32, 71–84.
- Ceyzériat, K.; Ben Haim, L.; Denizot, A.; Pommier, D.; Matos, M.; Guillemaud, O.; Palomares, M.A.; Abjean, L.; Petit, F.; Gipchtein, P.; et al. Modulation of Astrocyte Reactivity Improves Functional Deficits in Mouse Models of Alzheimer’s Disease. Acta Neuropathol. Commun. 2018, 6, 104.
- Haim, L.B.; Ceyzériat, K.; de Sauvage, M.A.C.; Aubry, F.; Auregan, G.; Guillermier, M.; Ruiz, M.; Petit, F.; Houitte, D.; Faivre, E.; et al. The JAK/STAT3 Pathway Is a Common Inducer of Astrocyte Reactivity in Alzheimer’s and Huntington’s Diseases. J. Neurosci. 2015, 35, 2817–2829.
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct Transfer of α-Synuclein from Neuron to Astroglia Causes Inflammatory Responses in Synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272.
- Wang, P.; Ye, Y. Filamentous Recombinant Human Tau Activates Primary Astrocytes via an Integrin Receptor Complex. Nat. Commun. 2021, 12, 95.
- Liu, X.; Ying, J.; Wang, X.; Zheng, Q.; Zhao, T.; Yoon, S.; Yu, W.; Yang, D.; Fang, Y.; Hua, F. Astrocytes in Neural Circuits: Key Factors in Synaptic Regulation and Potential Targets for Neurodevelopmental Disorders. Front. Mol. Neurosci. 2021, 14, 729273.
- Verkhratsky, A.; Nedergaard, M. Physiology of astroglia. Physiol. Rev. 2018, 98, 239–389.
- Panatier, A.; Vallée, J.; Haber, M.; Murai, K.K.; Lacaille, J.C.; Robitaille, R. Astrocytes Are Endogenous Regulators of Basal Transmission at Central Synapses. Cell 2011, 146, 785–798.
- Perea, G.; Araque, A. Properties of Synaptically Evoked Astrocyte Calcium Signal Reveal Synaptic Information Processing by Astrocytes. J. Neurosci. 2005, 25, 2192–2203.
- Clarke, B.E.; Taha, D.M.; Tyzack, G.E.; Patani, R. Regionally Encoded Functional Heterogeneity of Astrocytes in Health and Disease: A Perspective. Glia 2021, 69, 20–27.
- Delekate, A.; Füchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 Receptor Signalling Mediates Astrocytic Hyperactivity in Vivo in an Alzheimer’s Disease Mouse Model. Nat. Commun. 2014, 5, 5422.
- Reichenbach, N.; Delekate, A.; Breithausen, B.; Keppler, K.; Poll, S.; Schulte, T.; Peter, J.; Plescher, M.; Hansen, J.N.; Blank, N.; et al. P2Y1 Receptor Blockade Normalizes Network Dysfunction and Cognition in an Alzheimer’s Disease Model. J. Exp. Med. 2018, 215, 1649–1663.
- Harada, K.; Kamiya, T.; Tsuboi, T. Gliotransmitter Release from Astrocytes: Functional, Developmental, and Pathological Implications in the Brain. Front. Neurosci. 2016, 9, 499.
- Giaume, C.; Fromaget, C.; el Aoumari, A.; Cordier, J.; Glowinski, J.; Gros, D. Gap Junctions in Cultured Astrocytes: Single-Channel Currents and Characterization of Channel-Forming Protein. Neuron 1991, 6, 133–143.
- Dermietrel, R.; Hertzberg, E.L.; Kessler, J.A.; Spray, D.C. Gap Junctions between Cultured Astrocytes: Immunocytochemical, Molecular, and Electrophysiological Analysis. J. Neurosci. 1991, 11, 1421–1432.
- Batter, D.K.; Corpina, R.A.; Roy, C.; Spray, D.C.; Hertzberg, E.L.; Kessler, J.A. Heterogeneity in Gap Junction Expression in Astrocytes Cultured from Different Brain Regions. Glia 1992, 6, 213–221.
- Giaume, C.; Koulakoff, A.; Roux, L.; Holcman, D.; Rouach, N. Astroglial Networks: A Step Further in Neuroglial and Gliovascular Interactions. Nat. Rev. Neurosci. 2010, 11, 87–99.
- Heller, J.P.; Rusakov, D.A. Morphological Plasticity of Astroglia: Understanding Synaptic Microenvironment. Glia 2015, 63, 2133–2151.
- Henneberger, C.; Bard, L.; Panatier, A.; Reynolds, J.P.; Kopach, O.; Medvedev, N.I.; Minge, D.; Herde, M.K.; Anders, S.; Kraev, I.; et al. LTP Induction Boosts Glutamate Spillover by Driving Withdrawal of Perisynaptic Astroglia. Neuron 2020, 108, 919–936.
- Tani, H.; Dulla, C.G.; Farzampour, Z.; Taylor-Weiner, A.; Huguenard, J.R.; Reimer, R.J. A Local Glutamate-Glutamine Cycle Sustains Synaptic Excitatory Transmitter Release. Neuron 2014, 81, 888–900.
- Furness, D.N.; Dehnes, Y.; Akhtar, A.Q.; Rossi, D.J.; Hamann, M.; Grutle, N.J.; Gundersen, V.; Holmseth, S.; Lehre, K.P.; Ullensvang, K.; et al. A Quantitative Assessment of Glutamate Uptake into Hippocampal Synaptic Terminals and Astrocytes: New Insights into a Neuronal Role for Excitatory Amino Acid Transporter 2 (EAAT2). Neuroscience 2008, 157, 80–94.
- Danbolt, N.C.; Furness, D.N.; Zhou, Y. Neuronal vs Glial Glutamate Uptake: Resolving the Conundrum. Neurochem. Int. 2016, 98, 29–45.
- Roberts, R.C.; Roche, J.K.; McCullumsmith, R.E. Localization of Excitatory Amino Acid Transporters EAAT1 and EAAT2 in Human Postmortem Cortex: A Light and Electron Microscopic Study. Neuroscience 2014, 277, 522–540.
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of Glutamate Transporters Reveals a Major Role for Astroglial Transport in Excitotoxicity and Clearance of Glutamate. Neuron 1996, 16, 675–686.
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Aβ Induces Astrocytic Glutamate Release, Extrasynaptic NMDA Receptor Activation, and Synaptic Loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527.
- Scimemi, A.; Meabon, J.S.; Woltjer, R.L.; Sullivan, J.M.; Diamond, J.S.; Cook, D.G. Amyloid-Β1-42 Slows Clearance of Synaptically Released Glutamate by Mislocalizing Astrocytic GLT-1. J. Neurosci. 2013, 33, 5312–5318.
- Jacob, C.P.; Koutsilieri, E.; Bartl, J.; Neuen-Jacob, E.; Arzberger, T.; Zander, N.; Ravid, R.; Roggendorf, W.; Riederer, P.; Grünblatt, E. Alterations in Expression of Glutamatergic Transporters and Receptors in Sporadic Alzheimer’s Disease. J. Alzheimer’s Dis. 2007, 11, 97–116.
- Lauderback, C.M.; Hackett, J.M.; Huang, F.F.; Keller, J.N.; Szweda, L.I.; Markesbery, W.R.; Allan Butterfield, D. The Glial Glutamate Transporter, GLT-1, Is Oxidatively Modified by 4-Hydroxy-2-Nonenal in the Alzheimer’s Disease Brain: The Role of Aβ1-42. J. Neurochem. 2001, 78, 413–416.
- Masliah, E.; Alford, M.; DeTeresa, R.; Mallory, M.; Hansen, L. Deficient Glutamate Transport Is Associated with Neurodegeneration in Alzheimer’s Disease. Ann. Neurol. 1996, 40, 759–766.
- Salcedo, C.; Pozo Garcia, V.; García-Adán, B.; Ameen, A.O.; Gegelashvili, G.; Waagepetersen, H.S.; Freude, K.K.; Aldana, B.I. Increased Glucose Metabolism and Impaired Glutamate Transport in Human Astrocytes Are Potential Early Triggers of Abnormal Extracellular Glutamate Accumulation in HiPSC-Derived Models of Alzheimer’s Disease. J. Neurochem. 2023, 1–19.
- Zhang, Y.; Meng, X.; Jiao, Z.; Liu, Y.; Zhang, X.; Qu, S. Generation of a Novel Mouse Model of Parkinson’s Disease via Targeted Knockdown of Glutamate Transporter GLT-1 in the Substantia Nigra. ACS Chem. Neurosci. 2020, 11, 406–417.
- Iovino, L.; Giusti, V.; Pischedda, F.; Giusto, E.; Plotegher, N.; Marte, A.; Battisti, I.; Di Iacovo, A.; Marku, A.; Piccoli, G.; et al. Trafficking of the Glutamate Transporter Is Impaired in LRRK2-Related Parkinson’s Disease. Acta Neuropathol. 2022, 144, 81–106.
- Papadeas, S.T.; Kraig, S.E.; O’Banion, C.; Lepore, A.C.; Maragakis, N.J. Astrocytes Carrying the Superoxide Dismutase 1 (SOD1 G93A) Mutation Induce Wild-Type Motor Neuron Degeneration in Vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 17803–17808.
- Tong, J.; Huang, C.; Bi, F.; Wu, Q.; Huang, B.; Liu, X.; Li, F.; Zhou, H.; Xia, X.G. Expression of ALS-Linked TDP-43 Mutant in Astrocytes Causes Non-Cell-Autonomous Motor Neuron Death in Rats. EMBO J. 2013, 32, 1917–1926.
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-Linked SOD1 Mutant G85R Mediates Damage to Astrocytes and Promotes Rapidly Progressive Disease with SOD1-Containing Inclusions. Neuron 1997, 18, 327–338.
- Van Den Bosch, L.; Van Damme, P.; Bogaert, E.; Robberecht, W. The Role of Excitotoxicity in the Pathogenesis of Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2006, 1762, 1068–1082.
- Shin, J.Y.; Fang, Z.H.; Yu, Z.X.; Wang, C.E.; Li, S.H.; Li, X.J. Expression of Mutant Huntingtin in Glial Cells Contributes to Neuronal Excitotoxicity. J. Cell Biol. 2005, 171, 1001–1012.
- Faideau, M.; Kim, J.; Cormier, K.; Gilmore, R.; Welch, M.; Auregan, G.; Dufour, N.; Guillermier, M.; Brouillet, E.; Hantraye, P.; et al. In Vivo Expression of Polyglutamine-Expanded Huntingtin by Mouse Striatal Astrocytes Impairs Glutamate Transport: A Correlation with Huntington’s Disease Subjects. Hum. Mol. Genet. 2010, 19, 3053–3067.
- Jiang, R.; Diaz-Castro, B.; Looger, L.L.; Khakh, B.S. Dysfunctional Calcium and Glutamate Signaling in Striatal Astrocytes from Huntington’s Disease Model Mice. J. Neurosci. 2016, 36, 3453–3470.
- Wu, J.; Bie, B.; Foss, J.F.; Naguib, M. Amyloid Fibril–Induced Astrocytic Glutamate Transporter Disruption Contributes to Complement C1q-Mediated Microglial Pruning of Glutamatergic Synapses. Mol. Neurobiol. 2020, 57, 2290–2300.
- Villanueva, C.B.; Stephensen, H.J.T.; Mokso, R.; Benraiss, A.; Sporring, J.; Goldman, S.A. Astrocytic Engagement of the Corticostriatal Synaptic Cleft Is Disrupted in a Mouse Model of Huntington s Disease. Proc. Natl. Acad. Sci. USA 2023, 120, e2210719129.
- Andersen, J.V.; Christensen, S.K.; Westi, E.W.; Diaz-delCastillo, M.; Tanila, H.; Schousboe, A.; Aldana, B.I.; Waagepetersen, H.S. Deficient Astrocyte Metabolism Impairs Glutamine Synthesis and Neurotransmitter Homeostasis in a Mouse Model of Alzheimer’s Disease. Neurobiol. Dis. 2021, 148, 105198.
- Andersen, J.V.; Skotte, N.H.; Christensen, S.K.; Polli, F.S.; Shabani, M.; Markussen, K.H.; Haukedal, H.; Westi, E.W.; Diaz-delCastillo, M.; Sun, R.C.; et al. Hippocampal Disruptions of Synaptic and Astrocyte Metabolism Are Primary Events of Early Amyloid Pathology in the 5xFAD Mouse Model of Alzheimer’s Disease. Cell Death Dis. 2021, 12, 954.
- Salcedo, C.; Andersen, J.V.; Vinten, K.T.; Pinborg, L.H.; Waagepetersen, H.S.; Freude, K.K.; Aldana, B.I. Functional Metabolic Mapping Reveals Highly Active Branched-Chain Amino Acid Metabolism in Human Astrocytes, Which Is Impaired in IPSC-Derived Astrocytes in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 736580.
- Salcedo, C.; Wagner, A.; Andersen, J.V.; Vinten, K.T.; Waagepetersen, H.S.; Schousboe, A.; Freude, K.K.; Aldana, B.I. Downregulation of GABA Transporter 3 (GAT3) Is Associated with Deficient Oxidative GABA Metabolism in Human Induced Pluripotent Stem Cell-Derived Astrocytes in Alzheimer’s Disease. Neurochem. Res. 2021, 46, 2676–2686.
- Paumier, A.; Boisseau, S.; Jacquier-Sarlin, M.; Pernet-Gallay, K.; Buisson, A.; Albrieux, M. Astrocyte-Neuron Interplay Is Critical for Alzheimer’s Disease Pathogenesis and Is Rescued by TRPA1 Channel Blockade. Brain 2022, 145, 388–405.
- Trudler, D.; Sanz-Blasco, S.; Eisele, Y.S.; Ghatak, S.; Bodhinathan, K.; Akhtar, M.W.; Lynch, W.P.; Pinã-Crespo, J.C.; Talantova, M.; Kelly, J.W.; et al. A-Synuclein Oligomers Induce Glutamate Release from Astrocytes and Excessive Extrasynaptic NMDAR Activity in Neurons, Thus Contributing to Synapse Loss. J. Neurosci. 2021, 41, 2264–2273.
- Balu, D.T.; Pantazopoulos, H.; Huang, C.C.Y.; Muszynski, K.; Harvey, T.L.; Uno, Y.; Rorabaugh, J.M.; Galloway, C.R.; Botz-Zapp, C.; Berretta, S.; et al. Neurotoxic Astrocytes Express the D-Serine Synthesizing Enzyme, Serine Racemase, in Alzheimer’s Disease. Neurobiol. Dis. 2019, 130, 104511.
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from Reactive Astrocytes Impairs Memory in Mouse Models of Alzheimer’s Disease. Nat. Med. 2014, 20, 886–896.
- Shrivastava, A.N.; Kowalewski, J.M.; Renner, M.; Bousset, L.; Koulakoff, A.; Melki, R.; Giaume, C.; Triller, A. β-Amyloid and ATP-Induced Diffusional Trapping of Astrocyte and Neuronal Metabotropic Glutamate Type-5 Receptors. Glia 2013, 61, 1673–1686.
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.A.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFκB-Activated Astroglial Release of Complement C3 Compromises Neuronal Morphology and Function Associated with Alzheimer’s Disease. Neuron 2015, 85, 101–115.
- Sibille, J.; Pannasch, U.; Rouach, N. Astroglial Potassium Clearance Contributes to Short-Term Plasticity of Synaptically Evoked Currents at the Tripartite Synapse. J. Physiol. 2014, 592, 87–102.
- Bellot-Saez, A.; Kékesi, O.; Morley, J.W.; Buskila, Y. Astrocytic Modulation of Neuronal Excitability through K + Spatial Buffering. Neurosci. Biobehav. Rev. 2017, 77, 87–97.
- Tong, X.; Ao, Y.; Faas, G.C.; Nwaobi, S.E.; Xu, J.; Haustein, M.D.; Anderson, M.A.; Mody, I.; Olsen, M.L.; Sofroniew, M.V.; et al. Astrocyte Kir4.1 Ion Channel Deficits Contribute to Neuronal Dysfunction in Huntington’s Disease Model Mice. Nat. Neurosci. 2014, 17, 694–703.
- Kelley, K.W.; Ben Haim, L.; Schirmer, L.; Tyzack, G.E.; Tolman, M.; Miller, J.G.; Tsai, H.H.; Chang, S.M.; Molofsky, A.V.; Yang, Y.; et al. Kir4.1-Dependent Astrocyte-Fast Motor Neuron Interactions Are Required for Peak Strength. Neuron 2018, 98, 306–319.
- Galea, E.; Weinstock, L.D.; Larramona-Arcas, R.; Pybus, A.F.; Giménez-Llort, L.; Escartin, C.; Wood, L.B. Multi-Transcriptomic Analysis Points to Early Organelle Dysfunction in Human Astrocytes in Alzheimer’s Disease. Neurobiol. Dis. 2022, 166, 105655.
- Sonninen, T.M.; Hämäläinen, R.H.; Koskuvi, M.; Oksanen, M.; Shakirzyanova, A.; Wojciechowski, S.; Puttonen, K.; Naumenko, N.; Goldsteins, G.; Laham-Karam, N.; et al. Metabolic Alterations in Parkinson’s Disease Astrocytes. Sci. Rep. 2020, 10, 14474.
- Lindström, V.; Gustafsson, G.; Sanders, L.H.; Howlett, E.H.; Sigvardson, J.; Kasrayan, A.; Ingelsson, M.; Bergström, J.; Erlandsson, A. Extensive Uptake of α-Synuclein Oligomers in Astrocytes Results in Sustained Intracellular Deposits and Mitochondrial Damage. Mol. Cell. Neurosci. 2017, 82, 143–156.
- Rostami, J.; Holmqvist, S.; Lindström, V.; Sigvardson, J.; Westermark, G.T.; Ingelsson, M.; Bergström, J.; Roybon, L.; Erlandsson, A. Human Astrocytes Transfer Aggregated Alpha-Synuclein via Tunneling Nanotubes. J. Neurosci. 2017, 37, 11835–11853.
- Cassina, P.; Cassina, A.; Pehar, M.; Castellanos, R.; Gandelman, M.; De León, A.; Robinson, K.M.; Mason, R.P.; Beckman, J.S.; Barbeito, L.; et al. Mitochondrial Dysfunction in SOD1G93A-Bearing Astrocytes Promotes Motor Neuron Degeneration: Prevention by Mitochondrial-Targeted Antioxidants. J. Neurosci. 2008, 28, 4115–4122.
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human IPSC-Derived Astrocytes from ALS Patients with Mutated C9ORF72 Show Increased Oxidative Stress and Neurotoxicity. EBioMedicine 2019, 50, 274–289.
- Jiwaji, Z.; Hardingham, G.E. The Consequences of Neurodegenerative Disease on Neuron-Astrocyte Metabolic and Redox Interactions. Neurobiol. Dis. 2023, 185, 106255.
- Jiwaji, Z.; Tiwari, S.S.; Avilés-Reyes, R.X.; Hooley, M.; Hampton, D.; Torvell, M.; Johnson, D.A.; McQueen, J.; Baxter, P.; Sabari-Sankar, K.; et al. Reactive Astrocytes Acquire Neuroprotective as Well as Deleterious Signatures in Response to Tau and Aß Pathology. Nat. Commun. 2022, 13, 135.
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 Activation in Astrocytes Protects against Neurodegeneration in Mouse Models of Familial Amyotrophic Lateral Sclerosis. J. Neurosci. 2008, 28, 13574–13581.
- Gan, L.; Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Astrocyte-Specific Overexpression of Nrf2 Delays Motor Pathology and Synuclein Aggregation throughout the CNS in the Alpha-Synuclein Mutant (A53T) Mouse Model. J. Neurosci. 2012, 32, 17775–17787.
- SantaCruz, K.S.; Yazlovitskaya, E.; Collins, J.; Johnson, J.; DeCarli, C. Regional NAD(P)H:Quinone Oxidoreductase Activity in Alzheimer’s Disease. Neurobiol. Aging 2004, 25, 63–69.
- Magistretti, P.J.; Allaman, I. Lactate in the Brain: From Metabolic End-Product to Signalling Molecule. Nat. Rev. Neurosci. 2018, 19, 235–249.
- Figley, C.R. Lactate Transport and Metabolism in the Human Brain: Implications for the Astrocyte-Neuron Lactate Shuttle Hypothesis. J. Neurosci. 2011, 31, 4768–4770.
- Madji Hounoum, B.; Mavel, S.; Coque, E.; Patin, F.; Vourc’h, P.; Marouillat, S.; Nadal-Desbarats, L.; Emond, P.; Corcia, P.; Andres, C.R.; et al. Wildtype Motoneurons, ALS-Linked SOD1 Mutation and Glutamate Profoundly Modify Astrocyte Metabolism and Lactate Shuttling. Glia 2017, 65, 592–605.
- Ferraiuolo, L.; Higginbottom, A.; Heath, P.R.; Barber, S.; Greenald, D.; Kirby, J.; Shaw, P.J. Dysregulation of Astrocyte-Motoneuron Cross-Talk in Mutant Superoxide Dismutase 1-Related Amyotrophic Lateral Sclerosis. Brain 2011, 134, 2627–2641.
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, Š.; Gubert Olivé, M.; Shakirzyanova, A.; Leskelä, S.; Sarajärvi, T.; Viitanen, M.; et al. PSEN1 Mutant IPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem Cell Rep. 2017, 9, 1885–1897.
- Brandebura, A.N.; Paumier, A.; Onur, T.S.; Allen, N.J. Astrocyte Contribution to Dysfunction, Risk and Progression in Neurodegenerative Disorders. Nat. Rev. Neurosci. 2023, 24, 23–39.
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-Scale Proteomic Analysis of Alzheimer’s Disease Brain and Cerebrospinal Fluid Reveals Early Changes in Energy Metabolism Associated with Microglia and Astrocyte Activation. Nat. Med. 2020, 26, 769–780.
- Allen, S.P.; Hall, B.; Castelli, L.M.; Francis, L.; Woof, R.; Siskos, A.P.; Kouloura, E.; Gray, E.; Thompson, A.G.; Talbot, K.; et al. Astrocyte Adenosine Deaminase Loss Increases Motor Neuron Toxicity in Amyotrophic Lateral Sclerosis. Brain 2019, 142, 586–605.
- Allen, S.P.; Hall, B.; Woof, R.; Francis, L.; Gatto, N.; Shaw, A.C.; Myszczynska, M.; Hemingway, J.; Coldicott, I.; Willcock, A.; et al. C9orf72 Expansion within Astrocytes Reduces Metabolic Flexibility in Amyotrophic Lateral Sclerosis. Brain 2019, 142, 3771–3790.
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D.; Bryan Alzheimer, K. Apolipoprotein E: High-Avidity Binding to B-Amyloid and Increased Frequency of Type 4 Allele in Late-Onset Familial Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981.
- Kim, J.; Basak, J.M.; Holtzman, D.M. The Role of Apolipoprotein E in Alzheimer’s Disease. Neuron 2009, 63, 287–303.
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-Wide Meta-Analysis Identifies New Loci and Functional Pathways Influencing Alzheimer’s Disease Risk. Nat. Genet. 2019, 51, 404–413.
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 Markedly Exacerbates Tau-Mediated Neurodegeneration in a Mouse Model of Tauopathy. Nature 2017, 549, 523–527.
- Larramona-Arcas, R.; González-Arias, C.; Perea, G.; Gutiérrez, A.; Vitorica, J.; García-Barrera, T.; Gómez-Ariza, J.L.; Pascua-Maestro, R.; Ganfornina, M.D.; Kara, E.; et al. Sex-Dependent Calcium Hyperactivity Due to Lysosomal-Related Dysfunction in Astrocytes from APOE4 versus APOE3 Gene Targeted Replacement Mice. Mol. Neurodegener. 2020, 15, 35.
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Correlations between Apolipoprotein E Ε4 Gene Dose and Brain-Imaging Measurements of Regional Hypometabolism. Proc. Natl. Acad. Sci. USA 2005, 102, 8299–8302.
- Williams, H.C.; Farmer, B.C.; Piron, M.A.; Walsh, A.E.; Bruntz, R.C.; Gentry, M.S.; Sun, R.C.; Johnson, L.A. APOE Alters Glucose Flux through Central Carbon Pathways in Astrocytes. Neurobiol. Dis. 2020, 136, 104742.
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human IPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e7.
- TCW, J.; Qian, L.; Pipalia, N.H.; Chao, M.J.; Liang, S.A.; Shi, Y.; Jain, B.R.; Bertelsen, S.E.; Kapoor, M.; Marcora, E.; et al. Cholesterol and Matrisome Pathways Dysregulated in Astrocytes and Microglia. Cell 2022, 185, 2213–2233.e25.
- Al-Dalahmah, O.; Sosunov, A.A.; Shaik, A.; Ofori, K.; Liu, Y.; Vonsattel, J.P.; Adorjan, I.; Menon, V.; Goldman, J.E. Single-Nucleus RNA-Seq Identifies Huntington Disease Astrocyte States. Acta Neuropathol. Commun. 2020, 8, 19.
- Valenza, M.; Marullo, M.; Di Paolo, E.; Cesana, E.; Zuccato, C.; Biella, G.; Cattaneo, E. Disruption of Astrocyte-Neuron Cholesterol Cross Talk Affects Neuronal Function in Huntington’s Disease. Cell Death Differ. 2015, 22, 690–702.
- Birolini, G.; Verlengia, G.; Talpo, F.; Maniezzi, C.; Zentilin, L.; Giacca, M.; Conforti, P.; Cordiglieri, C.; Caccia, C.; Leoni, V.; et al. SREBP2 Gene Therapy Targeting Striatal Astrocytes Ameliorates Huntington’s Disease Phenotypes. Brain 2021, 144, 3175–3190.
- Kim, J.M.; Cha, S.H.; Choi, Y.R.; Jou, I.; Joe, E.H.; Park, S.M. DJ-1 Deficiency Impairs Glutamate Uptake into Astrocytes via the Regulation of Flotillin-1 and Caveolin-1 Expression. Sci. Rep. 2016, 6, 28823.
- Kim, K.S.; Kim, J.S.; Park, J.Y.; Suh, Y.H.; Jou, I.; Joe, E.H.; Park, S.M. DJ-1 Associates with Lipid Rafts by Palmitoylation and Regulates Lipid Rafts-Dependent Endocytosis in Astrocytes. Hum. Mol. Genet. 2013, 22, 4805–4817.
- Guttenplan, K.A.; Weigel, M.K.; Prakash, P.; Wijewardhane, P.R.; Hasel, P.; Rufen-Blanchette, U.; Münch, A.E.; Blum, J.A.; Fine, J.; Neal, M.C.; et al. Neurotoxic Reactive Astrocytes Induce Cell Death via Saturated Lipids. Nature 2021, 599, 102–107.
- Nagelhus, E.A.; Horio, Y.; Inanobe, A.; Fujita, A.; Haug, F.M.; Nielsen, S.; Kuhachi, Y.; Ottersen, O.P. Immunogold Evidence Suggests That Coupling of K+ Siphoning and Water Transport in Rat Retinal Muller Cells Is Mediated by a Coenrichment of Kir4.1 and AQP4 in Specific Membrane Domains. Glia 1999, 26, 47–54.
- Nagelhus, E.A.; Mathiisen, T.M.; Ottersen, O.P. Aquaporin-4 in the Central Nervous System: Cellular and Subcellular Distribution and Coexpression with KIR4.1. Neuroscience 2004, 129, 905–913.
- Amiry-Moghaddam, M.; Williamson, A.; Palomba, M.; Eid, T.; de Lanerolle, N.C.; Nagelhus, E.A.; Adams, M.E.; Froehner, S.C.; Agre, P.; Ottersen, O.P. Delayed K+ Clearance Associated with Aquaporin-4 Mislocalization: Phenotypic Defects in Brains of α-Syntrophin-Null Mice. Proc. Natl. Acad. Sci. USA 2003, 100, 13615–13620.
- Caron, N.S.; Banos, R.; Yanick, C.; Aly, A.E.; Byrne, L.M.; Smith, E.D.; Xie, Y.; Smith, S.E.P.; Potluri, N.; Black, H.F.; et al. Mutant Huntingtin Is Cleared from the Brain via Active Mechanisms in Huntington Disease. J. Neurosci. 2021, 41, 780–796.
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111.
- Xu, Z.; Xiao, N.; Chen, Y.; Huang, H.; Marshall, C.; Gao, J.; Cai, Z.; Wu, T.; Hu, G.; Xiao, M. Deletion of Aquaporin-4 in APP/PS1 Mice Exacerbates Brain Aβ Accumulation and Memory Deficits. Mol. Neurodegener. 2015, 10, 58.
- Hoshi, A.; Yamamoto, T.; Shimizu, K.; Ugawa, Y.; Nishizawa, M.; Takahashi, H.; Kakita, A. Characteristics of Aquaporin Expression Surrounding Senile Plaques and Cerebral Amyloid Angiopathy in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2012, 71, 750–759.
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human ApoE Isoforms Differentially Regulate Brain Amyloid-β Peptide Clearance. Sci. Transl. Med. 2011, 3, 89ra57.
- Streubel-Gallasch, L.; Giusti, V.; Sandre, M.; Tessari, I.; Plotegher, N.; Giusto, E.; Masato, A.; Iovino, L.; Battisti, I.; Arrigoni, G.; et al. Parkinson’s Disease-Associated LRRK2 Interferes with Astrocyte-Mediated Alpha-Synuclein Clearance. Mol. Neurobiol. 2021, 58, 3119–3140.
- Loria, F.; Vargas, J.Y.; Bousset, L.; Syan, S.; Salles, A.; Melki, R.; Zurzolo, C. α-Synuclein Transfer between Neurons and Astrocytes Indicates That Astrocytes Play a Role in Degradation Rather than in Spreading. Acta Neuropathol. 2017, 134, 789–808.
- Dilsizoglu Senol, A.; Samarani, M.; Syan, S.; Guardia, C.M.; Nonaka, T.; Liv, N.; Latour-Lambert, P.; Hasegawa, M.; Klumperman, J.; Bonifacino, J.S.; et al. α-Synuclein Fibrils Subvert Lysosome Structure and Function for the Propagation of Protein Misfolding between Cells through Tunneling Nanotubes. PLoS. Biol. 2021, 19, e3001287.
- Mazzulli, J.R.; Xu, Y.-H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell 2011, 146, 37–52.
- Morrone Parfitt, G.; Coccia, E.; Goldman, C.; Whitney, K.; Reyes, R.; Sarrafha, L.; Nam, K.H.; Sohail, S.; Jones, D.R.; Crary, J.F.; et al. Disruption of Lysosomal Proteolysis in Astrocytes Facilitates Midbrain Organoid Proteostasis Failure in an Early-Onset Parkinson’s Disease Model. Nat. Commun. 2024, 15, 447.
- Perea, J.R.; López, E.; Diéz-Ballesteros, J.C.; Ávila, J.; Hernández, F.; Bolós, M. Extracellular Monomeric Tau Is Internalized by Astrocytes. Front. Neurosci. 2019, 13, 442.
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A Single-Cell Atlas of Entorhinal Cortex from Individuals with Alzheimer’s Disease Reveals Cell-Type-Specific Gene Expression Regulation. Nat. Neurosci. 2019, 22, 2087–2097.
- Rostami, J.; Mothes, T.; Kolahdouzan, M.; Eriksson, O.; Moslem, M.; Bergström, J.; Ingelsson, M.; O’Callaghan, P.; Healy, L.M.; Falk, A.; et al. Crosstalk between Astrocytes and Microglia Results in Increased Degradation of α-Synuclein and Amyloid-β Aggregates. J. Neuroinflammation 2021, 18, 124.
- Nakamura, S.; Yoshimori, T. New Insights into Autophagosome–Lysosome Fusion. J. Cell Sci. 2017, 130, 1209–1216.
- Kulkarni, A.; Dong, A.; Kulkarni, V.V.; Chen, J.; Laxton, O.; Anand, A.; Maday, S. Differential Regulation of Autophagy during Metabolic Stress in Astrocytes and Neurons. Autophagy 2020, 16, 1651–1667.
- Jäger, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role for Rab7 in Maturation of Late Autophagic Vacuoles. J. Cell Sci. 2004, 117, 4837–4848.
- Funayama, M.; Nishioka, K.; Li, Y.; Hattori, N. Molecular Genetics of Parkinson’s Disease: Contributions and Global Trends. J. Hum. Genet. 2023, 68, 125–130.
- Madureira, M.; Connor-Robson, N.; Wade-Martins, R. LRRK2: Autophagy and Lysosomal Activity. Front. Neurosci. 2020, 14, 498.
- di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific IPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229.
- Szebényi, K.; Wenger, L.M.D.; Sun, Y.; Dunn, A.W.E.; Limegrover, C.A.; Gibbons, G.M.; Conci, E.; Paulsen, O.; Mierau, S.B.; Balmus, G.; et al. Human ALS/FTD Brain Organoid Slice Cultures Display Distinct Early Astrocyte and Targetable Neuronal Pathology. Nat. Neurosci. 2021, 24, 1542–1554.
- Ahmad, A.; Patel, V.; Xiao, J.; Moshahid Khan, M. The Role of Neurovascular System in Neurodegenerative Diseases. Mol. Neurobiol. 2020, 57, 4373–4393.
- Liddelow, S.; Barres, B. SnapShot: Astrocytes in Health and Disease. Cell 2015, 162, 1170.e1.
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-Associated Astrocytes in Alzheimer’s Disease and Aging. Nat. Neurosci. 2020, 23, 701–706.
- Nicaise, C.; Soyfoo, M.S.; Authelet, M.; De Decker, R.; Bataveljic, D.; Delporte, C.; Pochet, R. Aquaporin-4 Overexpression in Rat ALS Model. Anat. Rec. 2009, 292, 207–213.
- Ouali Alami, N.; Schurr, C.; Olde Heuvel, F.; Tang, L.; Li, Q.; Tasdogan, A.; Kimbara, A.; Nettekoven, M.; Ottaviani, G.; Raposo, C.; et al. NF-κB Activation in Astrocytes Drives a Stage-specific Beneficial Neuroimmunological Response in ALS. EMBO J. 2018, 37, e98697.
- Garbuzova-Davis, S.; Haller, E.; Saporta, S.; Kolomey, I.; Nicosia, S.V.; Sanberg, P.R. Ultrastructure of Blood-Brain Barrier and Blood-Spinal Cord Barrier in SOD1 Mice Modeling ALS. Brain Res. 2007, 1157, 126–137.
- Garbuzova-Davis, S.; Saporta, S.; Haller, E.; Kolomey, I.; Bennett, S.P.; Potter, H.; Sanberg, P.R. Evidence of Compromised Blood-Spinal Cord Barrier in Early and Late Symptomatic SOD1 Mice Modeling ALS. PLoS ONE 2007, 2, e1205.
- Miyazaki, K.; Ohta, Y.; Nagai, M.; Morimoto, N.; Kurata, T.; Takehisa, Y.; Ikeda, Y.; Matsuura, T.; Abe, K. Disruption of Neurovascular Unit Prior to Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis. J. Neurosci. Res. 2011, 89, 718–728.
- Winkler, E.A.; Sengillo, J.D.; Sullivan, J.S.; Henkel, J.S.; Appel, S.H.; Zlokovic, B.V. Blood-Spinal Cord Barrier Breakdown and Pericyte Reductions in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2013, 125, 111–120.
- Halliday, M.R.; Rege, S.V.; Ma, Q.; Zhao, Z.; Miller, C.A.; Winkler, E.A.; Zlokovic, B.V. Accelerated Pericyte Degeneration and Blood-Brain Barrier Breakdown in Apolipoprotein E4 Carriers with Alzheimer’s Disease. J. Cereb. Blood Flow Metab. 2016, 36, 216–227.
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E Controls Cerebrovascular Integrity via Cyclophilin A. Nature 2012, 485, 512–516.
- Nishitsuji, K.; Hosono, T.; Nakamura, T.; Bu, G.; Michikawa, M. Apolipoprotein E Regulates the Integrity of Tight Junctions in an Isoform-Dependent Manner in an in Vitro Blood-Brain Barrier Model. J. Biol. Chem. 2011, 286, 17536–17542.
- Spampinato, S.F.; Merlo, S.; Fagone, E.; Fruciano, M.; Barbagallo, C.; Kanda, T.; Sano, Y.; Purrello, M.; Vancheri, C.; Ragusa, M.; et al. Astrocytes Modify Migration of Pbmcs Induced by β-Amyloid in a Blood-Brain Barrier in Vitro Model. Front. Cell Neurosci. 2019, 13, 337.
- Hsiao, H.Y.; Chen, Y.C.; Huang, C.H.; Chen, C.C.; Hsu, Y.H.; Chen, H.M.; Chiu, F.L.; Kuo, H.C.; Chang, C.; Chern, Y. Aberrant Astrocytes Impair Vascular Reactivity in Huntington Disease. Ann. Neurol. 2015, 78, 178–192.
- de Rus Jacquet, A.; Alpaugh, M.; Denis, H.L.; Tancredi, J.L.; Boutin, M.; Decaestecker, J.; Beauparlant, C.; Herrmann, L.; Saint-Pierre, M.; Parent, M.; et al. The Contribution of Inflammatory Astrocytes to BBB Impairments in a Brain-Chip Model of Parkinson’s Disease. Nat. Commun. 2023, 14, 3651.
- D’Ambrosi, N.; Apolloni, S. Fibrotic Scar in Neurodegenerative Diseases. Front. Immunol. 2020, 11, 1394.
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte Scar Formation Aids Central Nervous System Axon Regeneration. Nature 2016, 532, 195–200.
- Verkhratsky, A.; Butt, A.; Li, B.; Illes, P.; Zorec, R.; Semyanov, A.; Tang, Y.; Sofroniew, M.V. Astrocytes in Human Central Nervous System Diseases: A Frontier for New Therapies. Signal Transduct. Target Ther. 2023, 8, 396.
- Kamermans, A.; Planting, K.E.; Jalink, K.; van Horssen, J.; de Vries, H.E. Reactive Astrocytes in Multiple Sclerosis Impair Neuronal Outgrowth through TRPM7-Mediated Chondroitin Sulfate Proteoglycan Production. Glia 2019, 67, 68–77.
- Shijo, T.; Warita, H.; Suzuki, N.; Kitajima, Y.; Ikeda, K.; Akiyama, T.; Ono, H.; Mitsuzawa, S.; Nishiyama, A.; Izumi, R.; et al. Aberrant Astrocytic Expression of Chondroitin Sulfate Proteoglycan Receptors in a Rat Model of Amyotrophic Lateral Sclerosis. J. Neurosci. Res. 2018, 96, 222–233.
- Mizuno, H.; Warita, H.; Aoki, M.; Itoyama, Y. Accumulation of Chondroitin Sulfate Proteoglycans in the Microenvironment of Spinal Motor Neurons in Amyotrophic Lateral Sclerosis Transgenic Rats. J. Neurosci. Res. 2008, 86, 2512–2523.
- Peters, S.; Zitzelsperger, E.; Kuespert, S.; Iberl, S.; Heydn, R.; Johannesen, S.; Petri, S.; Aigner, L.; Thal, D.R.; Hermann, A.; et al. The TGF-β System as a Potential Pathogenic Player in Disease Modulation of Amyotrophic Lateral Sclerosis. Front. Neurol. 2017, 8, 669.
- Schachtrup, C.; Ryu, J.K.; Helmrick, M.J.; Vagena, E.; Galanakis, D.K.; Degen, J.L.; Margolis, R.U.; Akassoglou, K. Fibrinogen Triggers Astrocyte Scar Formation by Promoting the Availability of Active TGF-β after Vascular Damage. J. Neurosci. 2010, 30, 5843–5854.
- Gris, P.; Tighe, A.; Levin, D.; Sharma, R.; Brown, A. Transcriptional Regulation of Scar Gene Expression in Primary Astrocytes. Glia 2007, 55, 1145–1155.
- Lian, H.; Zheng, H. Signaling Pathways Regulating Neuron–Glia Interaction and Their Implications in Alzheimer’s Disease. J. Neurochem. 2016, 136, 475–491.
- Herrmann, J.E.; Imura, T.; Song, B.; Qi, J.; Ao, Y.; Nguyen, T.K.; Korsak, R.A.; Takeda, K.; Akira, S.; Sofroniew, M.V. STAT3 Is a Critical Regulator of Astrogliosis and Scar Formation after Spinal Cord Injury. J. Neurosci. 2008, 28, 7231–7243.