Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Fanny Huang and Version 1 by Lev G. Murokh.
Mitochondria are commonly perceived as “cellular power plants”. In addition to energy conversion, mitochondria play many other roles in cell operations. They act in calcium signaling, stress response, and stem cell regulation and also serve as general cellular signaling hubs. They regulate aging and control cell death (apoptosis). Mitochondria play a crucial role as a mediator between the ECM and the cell nucleus, and monitoring and controlling this relationship can be important for cancer prevention and treatment. Mitochondrial defects can include aberrated metabolism (the Warburg effect) or signaling dysfunction (reactive oxygen species or ROS production).
- mitochondria
- carcinogenesis
1. Introduction
A mitochondrion (plural: mitochondria) is an organelle with a size between 0.75 and 3 μm and is present in most eukaryotic cells. The mitochondrion is called the “powerhouse of the cell” [1] because it converts chemical energy from food into adenosine triphosphate (ATP), a molecular “energy currency” used in many cell functions throughout organisms. The energy conversion process in mitochondria starts in the cell matrix with the tricarboxylic acid cycle (TCA, also known as the Krebs cycle or the citric acid cycle), a series of chemical reactions that release stored energy through the oxidation of acetyl-coenzyme A molecules, which are derived from carbohydrates, fatty acids, and proteins. The TCA cycle reduces nicotinamide adenine dinucleotide (NAD+) to NADH, consumes acetate and water, and releases carbon dioxide. The NADH generated by the TCA cycle is supplied into the oxidative phosphorylation (OXPHOS) pathway, and it donates electrons to the mitochondrial electron transport chain. The machinery for OXPHOS is located in the inner mitochondrial membrane, where electron energy is used in proton-pumping complexes to create and maintain a proton population gradient. The resulting protonmotive force facilitates the mechanical rotation of the F0-F1 complex, producing ATP. The coupling of electron and proton transfer events to ATP production is at the center of the chemiosmotic theory proposed by Peter Mitchell [2], which earned him the Nobel Prize in 1978.
In the distant past, an ancestor of modern mitochondria was engulfed by the protocell and, rather than becoming digested, remained in a symbiotic relationship [3,4][3][4]. The importance of this event for the development of life on Earth can be illustrated in the 12 h scale shown in Figure 1. If life emerges at noon, it takes less than an hour to develop photosynthesis. Cyanobacteria appear at a quarter past two, and they need less than two hours to create an oxygen-rich atmosphere. Oxygenation is followed by a notable delay of more than six hours until plants emerge. Finally, with a vast acceleration of events, evolution goes from dinosaurs to mammals, primates, and Homo Sapiens, whose whole history takes just four seconds on this scale. It can be argued that this acceleration period is caused by mitochondria becoming fully operational, providing more energy for living organisms. In some sense, this development parallels the Industrial Revolution when the exponential growth of production was facilitated by coal mining, i.e., using a very condensed form of energy. Incidentally, mitochondria are fully functional without the host cell. They maintain their composition, organization, membrane potential, and ability to fuse [5]; they are fully competent for respiration and ATP synthesis [6], as well as for protein import [7]. The same cannot be said about the host organism, as it would not survive without mitochondria.
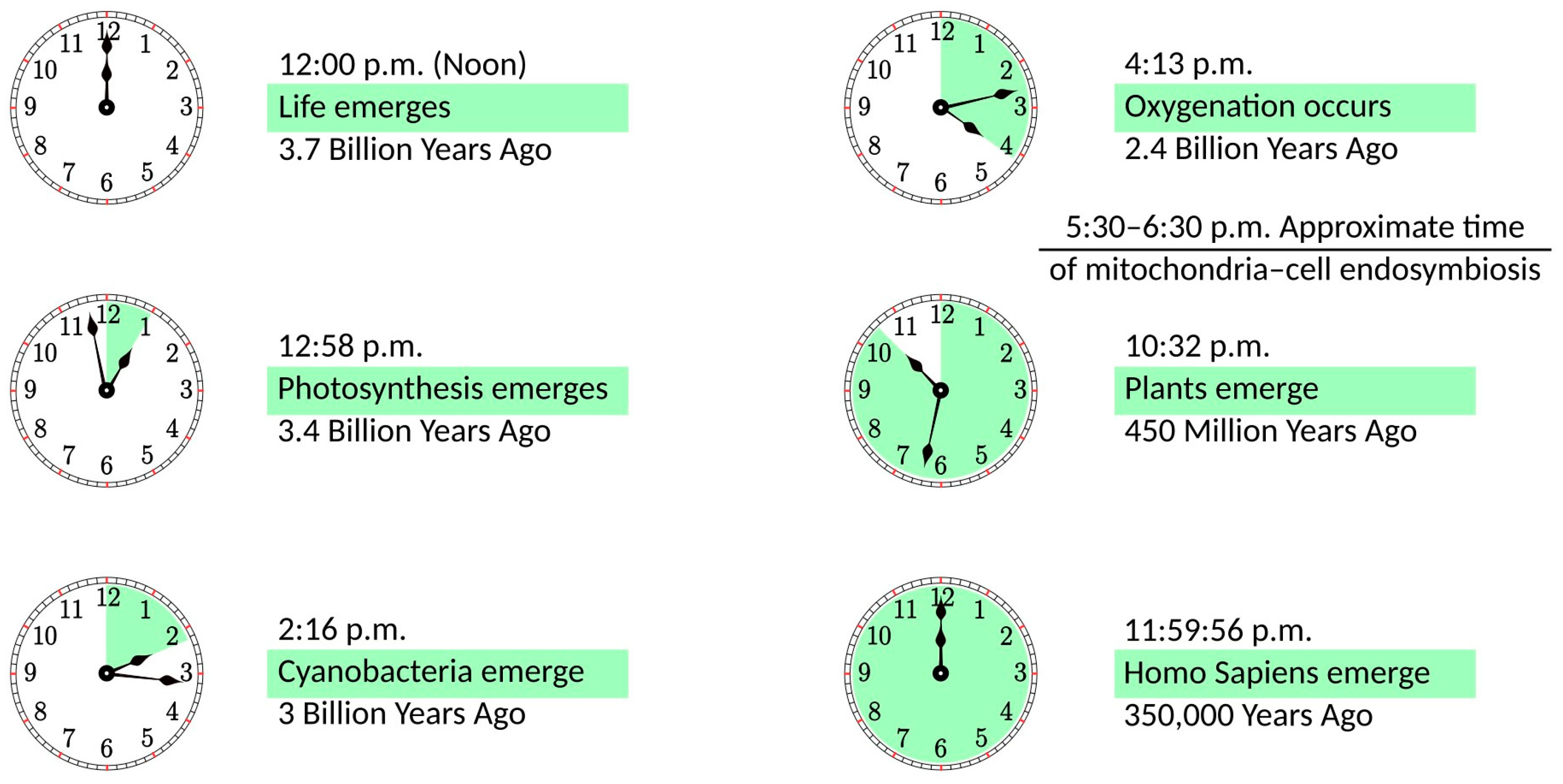
Figure 1.
The history of life on a 12 h scale, with life emerging at noon and the present time being at midnight.
In addition to energy conversion, mitochondria play many other roles in cell operations. They act in calcium signaling [8], stress response [9], and stem cell regulation [10,11][10][11] and also serve as general cellular signaling hubs [12]. They regulate aging [13] and control cell death (apoptosis) [14]. Given these functions, human health strongly depends on proper mitochondrial operation. In addition to specific mitochondrial disorders [15], there are links between mitochondrial dysfunction and many pathologies, including Parkinson’s, Alzheimer’s, and Huntington’s diseases [16]. Mitochondrial dysfunction is known to account for the development of most cardiovascular illnesses [17]. Even the resistance to SARS-CoV-2 is shown to be related to mitochondrial health [18].
2. Mitochondria and Carcinogenesis
2.1. General Discussion
Cancer is a major public health problem worldwide and the second leading cause of death in the United States, with an estimated 609,360 people dying from cancer in 2022, corresponding to almost 1700 deaths per day [19]. Billions of dollars have been spent on cancer-related research and trillions more on treatment. Still, despite certain improvements in cancer survival rates and the development of successful treatment methods for certain types of cancer, the War on Cancer is still far from victory. Diagnostics and treatments are primarily aimed at the latter stages when carcinogenesis becomes almost irreversible. To shift the paradigm, efforts should focus on precancerous changes, developing approaches to determine these changes and return to a healthy state.
Currently, the dominant concept of carcinogenesis is somatic mutation theory [20]. It states that a single “renegade cell” [21] acquires a set of sufficiently advantageous mutations that allows it to immortalize, proliferate autonomously, invade tissues, and metastasize. However, carcinogens cannot affect the cell’s nucleus directly, except for high-frequency irradiation. The immune system provides the initial response to carcinogens, and because of “overhealing” [22[22][23],23], the extracellular matrix (ECM) is altered, affecting bioelectric signaling [24]. The normal wound healing includes ECM remodeling and the formation of new tissues (scars). The overproduction of the cells and other factors involved in these processes, especially macrophages [23], can lead to overhealing and the start of carcinogenesis. The idea that ECM alteration is the origin of carcinogenesis was previously suggested [25[25][26],26], but the pathway from this starting point to DNA mutations was either ignored [25] or vaguely described [26]. Researchers believe that mitochondria play a crucial role as a mediator between the ECM and the cell nucleus, and monitoring and controlling this relationship can be important for cancer prevention and treatment. Mitochondrial defects can include aberrated metabolism (the Warburg effect) or signaling dysfunction (reactive oxygen species or ROS production).
2.2. Warburg Effect
Historically, the Warburg effect was mitochondria’s first and solid connection to carcinogenesis. In the presence of oxygen, mitochondria perform oxidative phosphorylation as the primary metabolic pathway. Alternatively, when oxygen is limited, anaerobic fermentation can metabolize glycolytic products, with lactate as a final product. The second pathway is much less effective, producing only two ATP molecules per glucose molecule, while OXPHOS can make 30 to 36 ATP molecules [27]. Over one hundred years ago, Otto Warburg observed that cancer cells use fermentation as the primary metabolic pathway, even in the presence of oxygen, and this process is called aerobic glycolysis [28,29][28][29]. He suggested that the associated mitochondrial dysfunction is the cause of cancer [30]. However, this hypothesis has been challenged in recent years due to findings that upregulated glycolysis in many cancers is not accompanied by detectable mitochondrial defects or OXPHOS disruptions [31,32][31][32]. In addition, there is evidence that the upregulation of glycolysis is not just for ATP synthesis but also for producing biomasses such as ribonucleotides [33] and amino acids [34].
With the general conclusion that cancer does not inactivate mitochondrial functions but instead alters its bioenergetic and biosynthetic state [35], the Warburg effect was studied concerning its role in cell signaling [36,37][36][37]. In particular, researchers uncovered its role in ROS regulation and redox balance [38[38][39],39], histone acetylation level [40[40][41],41], and oncogene-induced senescence [42]. However, in all cases, the Warburg effect is a consequence of tumorigenesis rather than its origin, as cancer cells actively reprogram their microenvironment.
2.3. ROS Production
The chemical reduction of O2 forms reactive oxygen species (ROS), including superoxide (O2−), hydrogen peroxide (H2O2), and the hydroxide (OH−). For many years, the perception of ROS was solely destructive, as they were associated with oxidative stress and thought to induce pathology generally by damaging lipids, proteins, and DNA [43]. Quite recently, it has become evident that ROS contribute to intracellular signaling to control numerous physiological and pathological cell processes [44,45][44][45]. The interplay of the damaging effects and regulatory functions of ROS has been discussed in several reviews [46,47,48][46][47][48].
The primary source of ROS within a cell is mitochondria [49]. The electron transport chain is a producer of ROS, with the electron leakages from Complexes I, II, and III creating O2− by single-electron oxygen reduction [50,51][50][51]. Under physiological conditions, it is estimated that 0.2 to 2% of leaked electrons do not contribute to ATP production [52]. While Complexes I and II exclusively create O2− in the mitochondrial matrix, Complex III produces O2− in both the matrix and intermembrane space [53,54][53][54]. In the latter case, O2− travels through voltage-dependent channels in the outer mitochondrial membrane and into the cytosol [55], where it can be converted into H2O2, participating in cellular signaling events [56].
Oxidative DNA damage can be a significant contributor to cancer [57]. In particular, the reaction of OH− with DNA accounts for most DNA strand breaks, representing the primary molecular reaction leading to carcinogenesis. To defend from the damage, cells use various antioxidant mechanisms, such as converting highly reactive O2− into H2O2 using superoxide dismutases [58]. However, a high concentration of H2O2 is also detrimental since H2O2 can be reduced to the damaging hydroxides OH− in the presence of Fe2+ and Cu2+ [59]. Producing the amount of ROS needed for self-signaling without harmful overproduction requires a delicate balance of various components, and its imbalance can mediate damage transfer from the ECM to DNA. One of the pathways to ROS overproduction can be aberrated bioelectric signaling in the ECM, increasing the mitochondrial membrane potential [60].
References
- Siekevitz, P. Powerhouse of the cell. Sci. Amer. 1957, 197, 131–140.
- Mitchell, P. Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic Type of Mechanism. Nature 1961, 191, 144–148.
- Margulis, L. Symbiosis in Cell Evolution; Freeman: San Francisco, CA, USA, 1981.
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial Evolution. Science 1999, 283, 1476–1481.
- Meeusen, S.; McCaffery, J.M.; Nunnari, J. Mitochondrial fusion intermediates revealed in vitro. Science 2004, 305, 1747–1752.
- Alexandre, A.; Reynafarje, B.; Lehninger, A.L. Stoichiometry of vectorial H+ movements coupled to electron transport and to ATP synthesis in mitochondria. Proc. Natl. Acad. Sci. USA 1978, 75, 5296–5300.
- Schmidt, O.; Pfanner, N.; Meisinger, C. Mitochondrial protein import: From proteomics to functional mechanisms. Nat. Rev. Mol. Cell Biol. 2010, 11, 655–667.
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signaling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578.
- Pellegrino, M.W.; Haynes, C.M. Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol. 2015, 13, 22.
- Lisowski, P.; Kannan, P.; Mlody, B.; Prigione, A. Mitochondria and the dynamic control of stem cell homeostasis. EMBO Rep. 2018, 19, e45432.
- Zhang, H.; Menzies, K.J.; Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 2018, 145, dev143420.
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34.
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957.
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118.
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080.
- Beal, M. Mitochondrial dysfunction in neurodegenerative diseases. Biochim. Biophys. Acta (BBA) Bioenerg. 1998, 1366, 211–223.
- Yang, J.; Guo, Q.; Feng, X.; Liu, Y.; Zhou, Y. Mitochondrial Dysfunction in Cardiovascular Diseases: Potential Targets for Treatment. Front. Cell Dev. Biol. 2022, 10, 841523.
- Nunn, A.V.W.; Guy, G.W.; Brysch, W.; Botchway, S.W.; Frasch, W.; Calabrese, E.J.; Bell, J.D. SARS-CoV-2 and mitochondrial health: Implications of lifestyle and ageing. Immun. Ageing 2020, 17, 33.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J Clin. 2022, 72, 7–33.
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724.
- Weinberg, R.A. One Renegade Cell: How Cancer Begins; Basic Books: New York, NY, USA, 1998.
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Eng. J. Med. 1986, 315, 1650–1659.
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628.
- Pietak, A.; Levin, M. Exploring Instructive Physiological Signaling with the Bioelectric Tissue Simulation Engine. Front. Bioeng. Biotech. 2016, 4, 55.
- Soto, A.M.; Sonnenschein, C. The Society of Cells: Cancer and Control of Cell Proliferation; Springer: New York, NY, USA, 1999.
- Brücher, B.L.D.M.; Jamall, I.S. Somatic Mutation Theory—Why it’s Wrong for Most Cancers. Cell. Physiol. Biochem. 2016, 38, 1663–1680.
- Hinkle, P.C. P/O ratios of mitochondrial oxidative phosphorylation. Biochim. Biophys. Acta (BBA) Bioenerg. 2005, 1706, 1–11.
- Warburg, O. The chemical constitution of respiration ferment. Science 1928, 68, 437–443.
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530.
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314.
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006, 9, 425–434.
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418.
- Tong, X.; Zhao, F.; Thompson, C.B. The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr. Opin. Genet. Dev. 2009, 19, 32–37.
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874.
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer. 2012, 12, 685–698.
- Locasale, J.W.; Cantley, L.C. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011, 14, 443–451.
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211.
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283.
- Hamanaka, R.B.; Chandel, N.S. Warburg effect and redox balance. Science 2011, 334, 1219–1220.
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080.
- Lu, C.; Thompson, C.B. Metabolic regulation of epigenetics. Cell Metab. 2012, 16, 9–17.
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.A.; Mackay, G.; Van Der Burg, S.H.; Verdegaal, E.M.; Cascante, M.; Shlomi, T.; et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 2013, 498, 109–112.
- Cross, C.E.; Halliwell, B.; Borish, E.T.; Pryor, W.A.; Ames, B.N.; Saul, R.L.; Mccord, J.M.; Harman, D. Oxygen radicals and human disease. Ann. Intern. Med. 1987, 107, 526–545.
- Weinberg, F.; Chandel, N.S. Reactive oxygen species-dependent signaling regulates cancer. Cell. Mol. Life Sci. 2009, 66, 3663–3673.
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883.
- Angelova, P.R.; Abramov, A.Y. Functional role of mitochondrial reactive oxygen species in physiology. Free Radic. Biol. Med. 2016, 100, 81–85.
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cell. Free Radic. Biol. Med. 2016, 100, 86–93.
- Brillo, V.; Chieregato, L.; Leanza, L.; Muccioli, S.; Costa, R. Mitochondrial Dynamics, ROS, and Cell Signaling: A Blended Overview. Life 2021, 11, 332.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13.
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312.
- Nickel, A.; Kohlhaas, M.; Maack, C. Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 2014, 73, 26–33.
- Zhao, R.; Jiang, S.; Zhang, L.; Yu, Z. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15.
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344.
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073.
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 2003, 278, 5557–5563.
- Orr, A.L.; Vargas, L.; Turk, C.N.; Baaten, J.E.; Matzen, J.T.; Dardov, V.J.; Attle, S.J.; Li, J.; Quackenbush, D.C.; Goncalves, R.L.; et al. Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 2015, 11, 834–836.
- Totter, J.R. Spontaneous cancer and its possible relationship to oxygen metabolism. Proc. Natl. Acad. Sci. USA 1980, 77, 1763–1767.
- Weisiger, R.A.; Fridovich, I. Mitochondrial superoxide dismutase: Site of synthesis and intramitochondrial localization. J. Biol. Chem. 1973, 248, 4793–4796.
- Winterbourn, C.C. The biological chemistry of hydrogen peroxide. Methods Enzymol. 2013, 528, 3–25.
- Kushnareva, Y.; Murphy, A.N.; Andreyev, A. I-mediated reactive oxygen species generation: Modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J. 2002, 368, 545–553.
More