Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Estefanía Lozano Velasco.
Effective intercellular communication is essential for cellular and tissue balance maintenance and response to challenges. Cellular communication methods involve direct cell contact or the release of biological molecules to cover short and long distances.
- intercellular communication
- extracellular vesicles
- cardiovascular diseases
1. Introduction
Intercellular communication is an essential activity for the maintenance of cellular and tissue homeostasis as well as responding to pathological processes. For this purpose, cells have different mechanisms depending on the distance of cell-to-cell communication requirements. Short cellular cross talk is mediated by direct cell-to-cell contact or secretion of soluble factors, whereas long range communication is driven through ligand–receptor interactions such as cytokine and/or hormone release [1,2][1][2]. However, in recent decades a novel mechanism of communication has emerged, where extracellular vesicles (EVs) are implicated [2]. EVs are constitutively produced by most of the cell types and consist in a lipidic bilayer membrane that encloses biological contents derived from the original cell, i.e., proteins, nucleic acids and lipids that can alter the biology of the distant cell [3,4][3][4]. The interest in EV biology has grown exponentially and many different types of EVs have been classified depending on their size, biogenesis and their biophysical properties [5,6][5][6] (Figure 1). There are two primary types of EVs, exosomes and ectosomes, whose classification is determined by their formation process. The genesis of these vesicles is contingent upon the assembly of local microdomains within endocytic membranes for exosomes and the plasma membrane for ectosomes, the latter subclassified into microvesicles and apoptotic bodies [7,8][7][8].
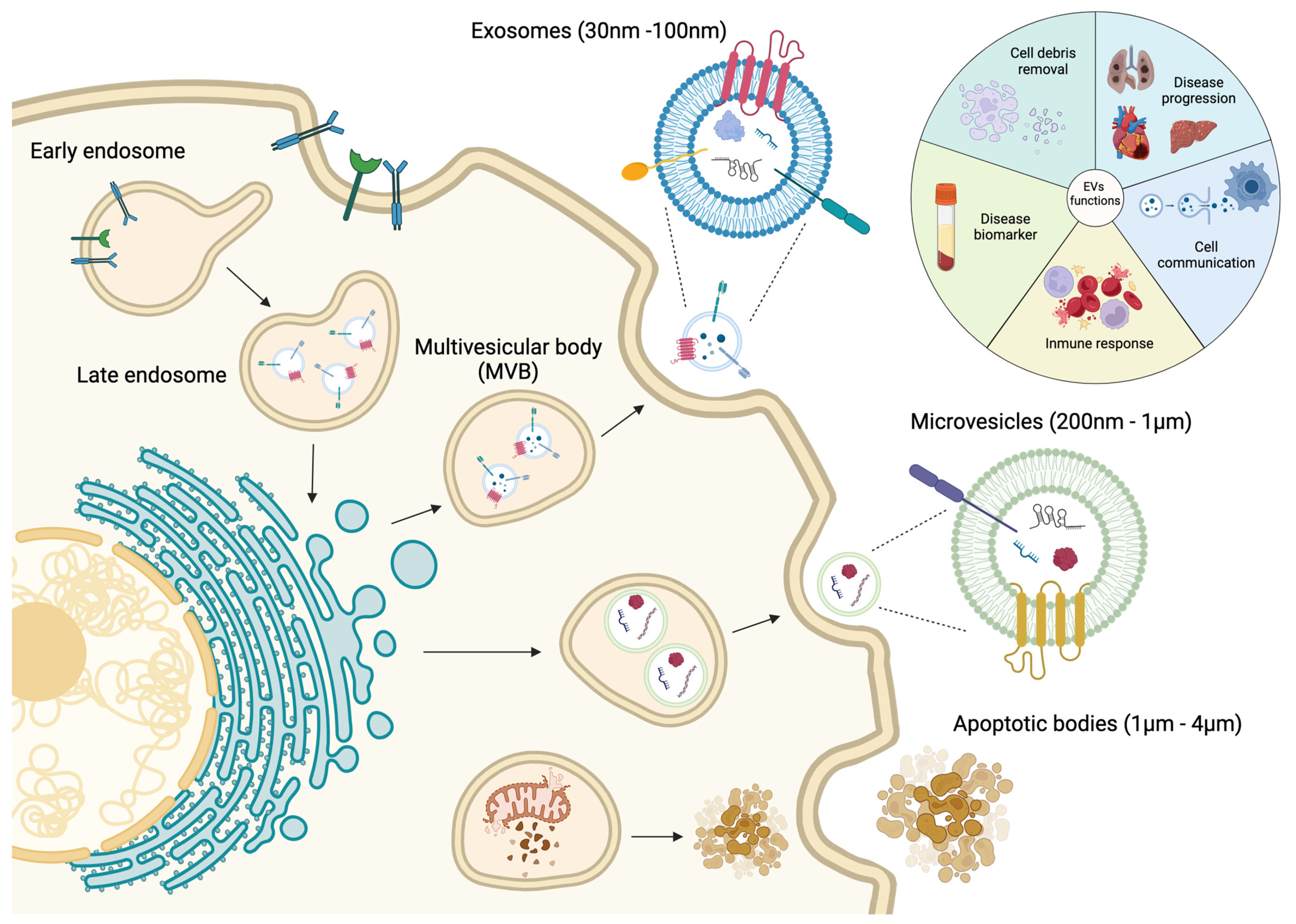
Figure 1. Schematic representation of the biological process of the genesis of extracellular vesicles, including the major classification due their size, content and functional impact.
Exosomes are small EVs ranging from 30 to 100 nm. They are formed by inward budding of the early endosome membrane which subsequently matures into multivesicular bodies (MVBs) [9,10,11,12][9][10][11][12]. MVBs are involved in protein sorting, recycling, storage, transport and exosome release into the extracellular space [11,13,14,15,16][11][13][14][15][16]. Exosomes were originally considered as a cellular mechanism to eliminate cell debris; however, since few years ago, they are considered as principal mediators of cell-to-cell communication, cell and tissue maintenance and disease progression [17]. Moreover, exosomes can act as antigen-presenting vesicles participating in the stimulation of immune response [18,19][18][19]. Microvesicles (MVs) are EVs with a range from 200 nm to 1 µm which are directly shed as an outward budding of the plasma membrane [10,11,12,13,20,21][10][11][12][13][20][21]. Similarly to exosomes, MVs are involved in cell-to-cell communication between healthy and diseased cells [22,23][22][23]. Finally, apoptotic bodies are released into the extracellular space as blebs of cells undergoing apoptosis, and they have been reported as the bigger EVs, with a size range from 1 to 4 µm [13,24][13][24]. These apoptotic bodies are formed when the cytoskeleton is separated from the cellular plasma membrane due to increased hydrostatic pressure after the cell contracts [25]. The composition of the apoptotic bodies are organelles, chromatin and glycosylated proteins, in contrast to exosomes and MVs [13,26,27][13][26][27] (Figure 1).
As mentioned previously, most cell types release EVs of different sizes, composition and subcellular origin, and moreover, these EVs can be found in different body fluids such as plasma, saliva and urine [28,29,30][28][29][30]. Therefore, due to their presence in liquid biopsies, EVs are considered as potential disease biomarkers. Understanding the mechanisms by which molecular cargos are loaded into the EVs will provide the key to completely understanding the role of EVs in cellular communication during disease development and progression [31,32,33,34][31][32][33][34] (Figure 1).
Cardiovascular disease (CVD) is one of the leading causes of morbidity and mortality around the world [35]. CVDs cover a wide range of pathological conditions where the most common types are ischemic heart disease, structural cardiomyopathies, electrical heart diseases and finally heart failure (HF) [36]. There have been major steps forward in diagnosis and prognosis of CVDs that have improved patients’ survival and their quality of life, but there is still much work to do for CVD prevention and palliation, given the continued high mortality rate [37]. Several studies have recently indicated that small EVs can be released by cardiovascular cells, such as cardiomyocytes (CM), endothelial cells (EC), cardiac fibroblasts (CF), platelets, smooth muscle cells (SMCs), leucocytes, monocytes and macrophages [38]. These small EVs play important biological roles in the maintenance of normal cardiac structure and function, whereas small EVs are able to change their composition under pathological conditions, thus contributing to the development of CVDs [39,40,41][39][40][41].
2. Small EVs in the Pathological Process of the Myocardial Infarction
Myocardial infarction (MI) is one of the most common CVDs, representing the main leading cause of death in the world [42]. Pathologically, MI is defined as the death of CMs due to a prolonged lack of oxygen in a specific area of the myocardium, initiating an apoptotic process leading to necrosis of the cardiac muscle and subsequently to other cardiac diseases such as arrhythmias or HF [43,44,45,46][43][44][45][46] (Figure 2A). The main etiology of MI is associated with the rupture of the atherosclerotic plate [47], although other causes, such as coronary artery embolism or coronary vasospasm have been also described [48]. After MI and the consequent death of CMs, autophagy and inflammatory processes begin as a strategy to remove the damaged tissue, allowing the replacement of the necrotic area with fibrotic tissue [46,49,50,51,52,53,54][46][49][50][51][52][53][54]. Furthermore, mechanisms of revascularization are also activated despite the low regenerative capacity of the heart after cardiac injury [55,56][55][56]. Currently, several studies show that exosomes play an important role in post-MI processes and participate in cellular communication regulating cardiac remodeling after MI [57,58,59][57][58][59]. For this reason, many authors focus their attention on the main mechanisms that regulate the production and content of these exosomes using transcriptomic and proteomic approaches, highlighting their detection as biomarkers after MI and thus their plausible therapeutic use [60,61,62][60][61][62]. The following paragraphs summarize the most important features of these exosomes in the MI context.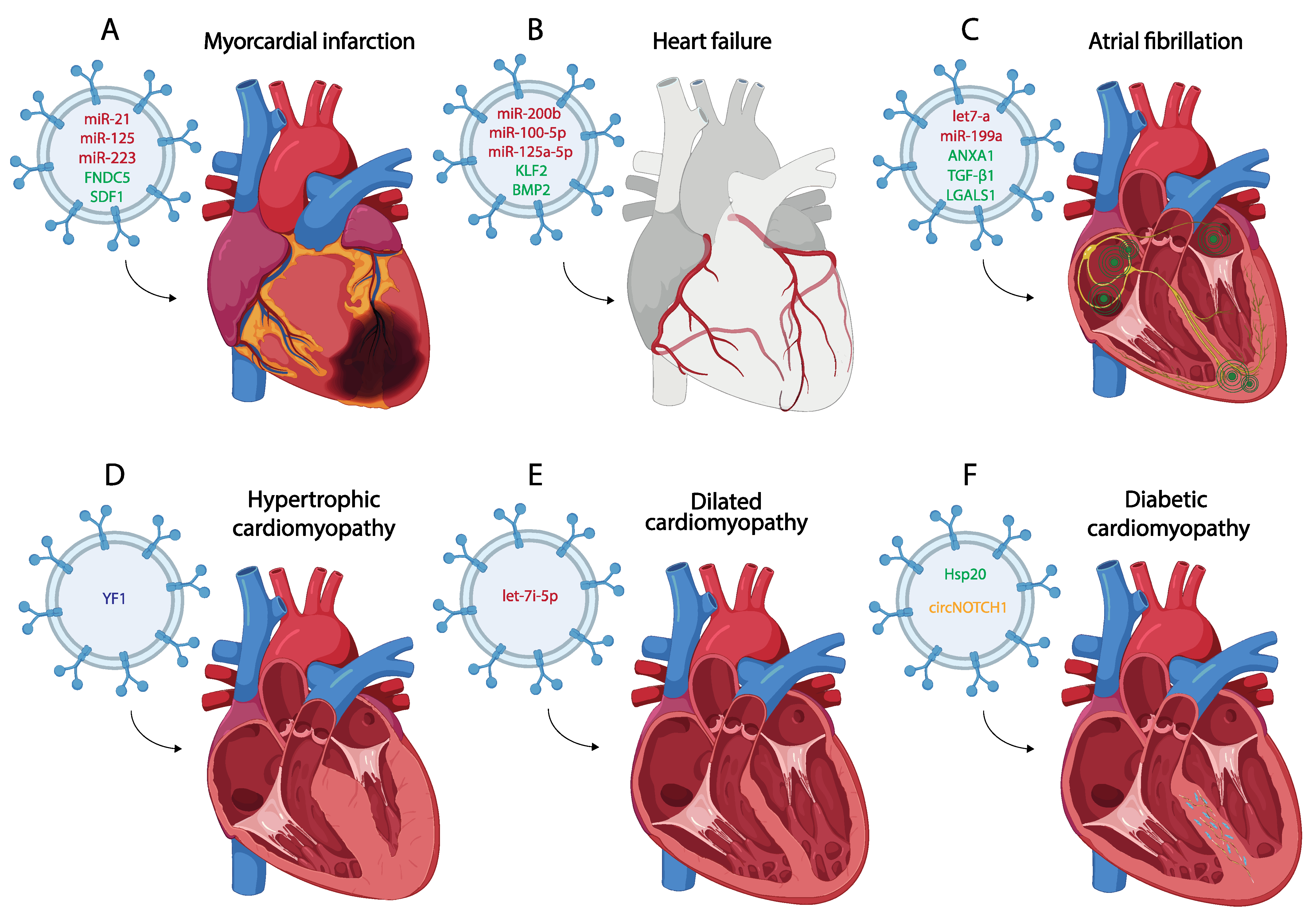
Figure 2. Schematic representation of the most representative small EV therapy in the treatment of different of myocardial infarction (A), heart failure (B), atrial fibrillation (C), hypertrophic cardiomyopathy (D), dilated cardiomyopathy (E) and diabetic cardiomyopathy (F). (miRNA red, protein green, lncRNA blue, circRNA yellow).
2.1. Small Extracellular Vesicle Transcriptomic Analyses in Myocardial Infarction
The discovery of new functions of exosomes has allowed the use of these small EVs as a potential tool to identify molecules that can be used as biomarkers after MI [63]. Currently, the main diagnostic biomarker for MI is cardiac troponin (T/I) [64], even though other more sensitive biomarkers have emerged, for example, non-coding RNAs, which allow for an earlier diagnosis [65,66][65][66]. Within non-coding RNAs, an increasing number of transcriptomic studies identified miRNAs in exosomes after MI [67,68,69][67][68][69]. Some analyses of circulating exosomes in the serum of MI patients have revealed a large number of miRNAs that are deregulated. For example, miR-203, miR-4516, or miR-183, which regulate the activity of several protein kinases in CMs, are upregulated, confirming their identification as MI biomarkers [67,70][67][70]. Another transcriptomic study identifies the deregulation of miRNA levels in exosomes on a large scale, identifying around 500 upregulated and downregulated miRNAs in MI exosomes, highlighting miR-6718 and miR-4329 [68]. Some miRNAs have been widely studied due to their essential role in post-MI inflammatory, fibrotic and angiogenic processes, i.e., miR-126 or miR-155, which are upregulated in exosomes of MI patients, and miR-21 or miR-146a-5p, which are downregulated [71,72,73][71][72][73]. Specifically, miR-146a-5p is associated with inflammation after MI by regulating M1 macrophage polarization through the regulation of TNF Receptor-Associated Factor 6 (TRAF6) [74]. Regarding angiogenesis, another transcriptomic analysis of exosomes derived from MI patients revealed up to 40 differentially expressed miRNAs, highlighting the downregulation of miR-143. In vitro assays demonstrated that this downregulation promotes angiogenesis through the insulin-like growth factor 1 receptor and nitric oxide (IGF-IR/NO) signaling pathway [75]. In contrast, in vitro assays in CMs after ischemia/reperfusion (I/R) describe exosomes with high levels of miR-143 and miR-222, both enhancing angiogenesis and cardiac remodeling [76]. In addition, in vitro and in vivo assays in necrotic dendritic cells revealed the presence of eight upregulated miRNAs related to the regulation of angiogenesis, in which miR-494 is the most significant due its role in promoting revascularization after injury [77]. The protective role of some exosomal miRNAs was also confirmed in ferroptosis after MI. Low levels of miR-26b-5p were described in exosomes derived from MI patients, which can reduce ferroptosis by positively regulating Solute Carrier Family 7 Member 11 (SLC7A11) [79][78]. Finally, other studies also associate miRNA transport in exosomes derived from MI patients with subsequent apoptosis [80][79]. Sun et al. (2022) revealed 52 differentially expressed miRNAs by transcriptomic analysis, highlighting the upregulation of miR-133a-3p, miR-151a-5p, miR-199b-5p, miR-374b-5p, miR-503-5p, and miR-708-5p. Although miRNAs are the most frequent non-coding RNAs studied in exosomes as MI biomarkers, long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs) are also analyzed. For lncRNAs, sequencing profiles allowed uresearchers to identify 518 differentially expressed in post-MI exosomes, highlighting ENST00000556899.1 and ENST00000575985.1, which are upregulated, thus supporting their involvement in the regulation of post-MI processes [81][80]. More specifically, other lncRNAs like TUG1 or HCG15 have also been detected as being upregulated in MI exosomes, and in vitro and in vivo assays show that these lncRNAs are implicated in the inhibition of angiogenesis and cell viability through the regulation of HIF-1α/VEGF-α and NF-κβ/p65, respectively [82,83][81][82]. Despite being smaller in number, circular RNAs have been reported to have a significant advantage over linear RNAs because they exhibit better stability and can therefore be identified as biomarkers of MI for a longer period of time [84][83]. For this reason, some transcriptomic studies focused on the analysis of circRNAs in MI exosomes, highlighting, for example, the upregulation of circ_0020887 and circ_0009590 in the exosomes patients with ST segment elevation on electrocardiogram [85][84]. Another circRNA that also increases its expression in exosomes after MI is circITGB1. Within in vivo assays using a mouse model, these exosomes activate dendritic cells and exacerbate cardiac damage and inflammation through the miR-342-3p/NFAM1 pathway [86][85]. In vitro assays have also been reported in CMs subjected to hypoxia where high levels of circ_HIPK3 and circ_SLC8A1 are detected [87,88][86][87]. These circ_HIPK3-loaded exosomes target cardiac microvascular endothelial cells (CMVECs), protecting them from oxidative stress through the regulation of the miR-29a/VEGFA and miR-33a-5p/IRS1 pathways [89,90][88][89].2.2. Small Extracellular Vesicle Proteomic Analyses in Myocardial Infarction
Just as many transcriptomic studies have identified essential biomarkers for MI; there are also proteomic analyses that reveal the presence of distinct proteins in post-MI exosomes that can be used as biomarkers for MI diagnosis [91][90]. Xie et al. (2022) analyzed the protein profile within the exosomes of post-MI patients, identifying 72 differentially expressed proteins. Notably, three proteins exhibited elevated levels: Plasminogen (PLG), Complement Component C8 Beta (C8B) and Thrombin (F2) [91][90]. Furthermore, additional research emphasizes the presence of proteins within these exosomes that are involved in the inflammatory process and cardiac remodeling, such as Phosphatase and Tensin Homolog (PTEN) or Matrix Metalloproteinase-9 (MMP-9) [71,92][71][91]. In the post-fibrotic process after MI, some proteins present in exosomes have also been described, such as the transcriptional cofactor Limb Bud And Heart Development Protein Homolog (LBH). These exosomes produced by damaged CMs are taken up by CFs and activate Crystallin Alpha B (CRYAB), promoting further proliferation of CFs and their differentiation into myofibroblasts [59]. Post-MI exosomes have also been reported to carry proteins that promote cardiac repair and remodeling after damage, for example, Clusterin or Profilin 2 (PFN2) [93,94][92][93]. The levels of PFN2 are elevated in exosomes produced by ECs following MI. In both in vitro and in vivo assays, these exosomes demonstrate an increase in angiogenesis and cardiac improvement after damage through regulation of the PI3K/PNF2/ERK axis [94][93]. Finally, other studies highlight the importance of exosomes as biomarkers for MI diagnosis. This research conducted a proteomic study by comparing plasma from control and MI patients and analyzing the protein profile in post-MI exosomes. These results revealed 11 proteins that were deregulated in the exosomes compared to control plasma. However, three of these proteins, Chymotrypsin C (CTRC), Proto-oncogene Tyrosine-protein Kinase SRC (SRC), and C-C Motif Chemokine Ligand 17 (CCL17), did not exhibit downregulation when comparing their levels in serum between MI and control patients. Therefore, this analysis justified the need to also analyze post-MI exosomes to obtain additional diagnostic information that are not available only from plasma samples [95][94].2.3. Mechanistic Insights into Small Extracellular Vesicle Related with Myocardial Infarction
As discussed in previous paragraphs, exosomes are involved in the regulation of the activation/inhibition of the main processes that occur after MI, such as inflammation, fibrosis, angiogenesis or apoptosis, as well as allowing communication between cells to coordinate these processes [57,58,59][57][58][59]. After MI, M1 macrophages are activated and increase the production of exosomes (M1-exos) with miR-155, which deliver this miRNA to CFs, thereby enhancing the inflammatory and fibrotic process [96][95]. M1-exos with high levels of miR-155 are also implicated in the inhibition of angiogenesis since these M1-exos can also be transferred to ECs and reduce angiogenesis via Sirt1/AMPKα2 and RAC1/PAK2 signaling after MI [97][96]. Macrophages are not the only cells that can release exosomes carrying miRNAs involved in post-MI process regulation. In vivo assays in a mouse MI model show that after MI, T CD4+ cells are activated and promote the synthesis of exosomes in which miR-142-3p is upregulated and promotes myofibroblast differentiation in ECs via miR-142-3p/APC/Wnt signaling [98][97]. Exosomes are involved in paracrine regulatory processes, as damaged CMs release exosomes after MI that will act on neighboring cells such as other CMs or ECs. This exosomal function is confirmed by the study of Gou et al. (2016), which showed that infarcted CMs generate exosomes that exhibit highly expressed miR-19a-3p, which are delivered to ECs and inhibit angiogenesis regulating Hypoxia-inducible factor 1-alpha (HIF-1α) [99][98]. Furthermore, post-MI CMs can generate exosomes after cardiac damage carrying miR-328-3p, which increase the apoptotic process through upregulation of Caspase 3 (Casp3) [100][99]. CFs also receive these exosomes produced by the CMs after MI, increasing their proliferation and the myofibroblast differentiation due to the high levels of miRNAs such us miR-208 or miR-92a [101,102,103][100][101][102]. Cell-to-cell communication via exosomes also takes place in the opposite direction, i.e., ECs can generate exosomes carrying miR-503 and promote CM apoptosis after MI [80][79]. Another example of miRNA that can be transferred between CMs after MI is miR-30a, whose levels are upregulated in exosomes extracted from the serum of MI patients. In vitro assays show that after hypoxia, these exosomes regulate the autophagy process between hypoxic CMs by upregulating genes such as Beclin-1 (BECN1), Autophagy Related 12 (Atg12), or Microtubule Associated Protein 1 Light Chain 3 Alpha (LC3I/II) [104][103]. Exosomes, carrying low levels of miR-342-3p as a consequence of MI, contribute to the regulation of both autophagy and apoptosis. Both in vitro and in vivo assays reveal that miR-342-3p regulates apoptosis and autophagy through the SRY-Box Transcription Factor 6 (SOX6) and Transcription Factor EB (TFEB), respectively [105][104]. A similar process, ferroptosis, also plays a relevant role in exosome regulation in post-MI processes. After MI, ferroptotic CMs release exosomes with low levels of miR-106b-3p, which promotes the activation of the Wnt pathway and increases the polarization of M1 macrophages, enhancing the inflammatory process after damage [106][105]. Other authors focused on the differential expression of proteins involved in the biogenesis, uptake or polarization of exosomes generated after MI. One example is the high level of CD44 in MI, which is involved in the synthesis of exosomes after MI through the positive regulation of Fibroblast Growth Factor Receptor 2 (FGFR2), as well as the subsequent uptake of these exosomes by ECs [107][106]. Regarding the ability of exosomes to migrate and the factors that trigger their polarization to the region of interest, it has been reported that dendritic cells increase the expression of C-C Motif Chemokine Receptor 7 (CCR7) after damage, and they generate exosomes containing both CCR7 and its ligands. These exosomes can target the spleen to activate CD4+ T cells, which produce anti-inflammatory cytokines that promote cardiac remodeling [108,109][107][108]. In addition, an external inflammatory stimulus, such as a decrease in the anti-inflammatory cytokine IL-10, can modify the protein content of exosomes. One protein that modifies its levels in exosomes is Integrin Linked Kinase (ILK), whose high levels promote the activation of NF-κβ, enhancing the inflammatory response and decreasing angiogenesis in the cells that receive these exosomes [110,111][109][110].2.4. Therapeutic Approaches
Although many studies describe the potential use of exosomes as biomarkers for the diagnosis of MI, several studies additionally analyzed the use of exosomes as tools to transport specific molecules that promote cardiac repair after MI, thus becoming a widely used strategy in recent years [112,113,114,115][111][112][113][114]. The most frequent strategy is based on the use of exosomes derived from different mesenchymal stem cells (MSCs) that carry a specific molecule (RNA or protein) involved in the processes of inflammation, fibrosis or angiogenesis, among others [116,117,118][115][116][117]. One of the most widely used miRNAs as a therapeutic tool due to its role in inflammation and fibrosis is miR-21. Several studies extract exosomes derived from different cell types such as MSCs, cardiac telocytes (CTs), or even serum from control individuals loaded with miR-21, using these exosomes as a treatment for MI in both in vitro and in vivo assays [119,120,121][118][119][120]. Administration of these miR-21-loaded exosomes shows an improvement in cardiac function after MI due to the positive regulation of the angiogenic process and the inhibition of CM apoptosis and fibrosis through the regulation of PTEN and p53/Cdip1/Casp3 signaling pathways, among others [119,121,122,123][118][120][121][122] (Figure 2A). MSC-derived exosomes, both under control and hypoxic conditions and carrying high levels of miR-125b, have also been used as a therapy against CM apoptosis after MI [124,125][123][124]. One of the most commonly used types of MSCs to obtain exosomes, administered as a treatment for MI, are those derived from human umbilical cord mesenchymal stem cells (HUCMSCs) [126,127][125][126]. These cells have been used to obtain exosomes that act as a vehicle to transport miRNAs such as miR-23, miR-133 or miR-223 to the infarcted area, which promote cardiac repair by activating angiogenesis and reducing inflammation, ferroptosis or fibrosis [128,129,130][127][128][129] (Figure 2). Other studies use this strategy to produce exosomes derived from cardiosphere-derived cells (CDCs) or MSCs which carry miR-181, regulating macrophage polarization and reducing inflammation when they are administered after MI [131,132][130][131]. There is also evidence that the therapeutic use of MSC-derived exosomes carrying specific proteins such as Itchy E3 Ubiquitin Protein Ligase (ITCH), Fibronectin Type III Domain Containing 5 (FNDC5) or Stromal Cell-Derived Factor 1 (SDF1) promote cardiac repair after their administration [133,134,135][132][133][134] (Figure 2A). Another treatment strategy post MI involves the modification of gene expression in the cells from which exosomes are subsequently extracted. This modification leads to changes in the content of these exosomes, offering a targeted approach for therapeutic interventions [136,137,138][135][136][137]. Several studies report that the overexpression of genes such as Hypoxia Inducible Factor 1 Subunit Alpha (HIF1-α) in MSCs results in a modification of the content of these exosomes, making them able to ameliorate cardiac damage after MI by promoting angiogenesis and reducing fibrosis, apoptosis or inflammation [139,140][138][139]. Overexpression of GATA-Binding Protein 4 (GATA4) in the cells from which exosomes are extracted has also been widely used to modify the exosome content for use as a treatment after MI [141,142,143][140][141][142]. He et al. (2018) revealed that overexpression of GATA4 in bone marrow mesenchymal stem cells (BMSCs) results in the production of exosomes that enhance myocyte precursor differentiation and reduce apoptosis after MI. This effect is due to a shift in the protein pattern carried by these exosomes, in which eight proteins associated with differentiation and six related to apoptosis have been identified with differential expression [141][140].3. Small EVs in the Pathological Process of Cardiomyopathies
Cardiomyopathies are defined as “a myocardial disorders in which the heart muscle is structurally and functionally abnormal, in the absence of coronary artery disease, hypertension, valvular disease, and congenital heart disease, however, sufficient to cause the observed myocardial abnormality” [207][143]. Cardiomyopathies can be classified into primaries or secondaries. Primary cardiomyopathies are mostly idiopathic, leading to heart failure and sudden death, while secondary cardiomyopathies develop in response to several extrinsic factors such as hypertension, metabolic disorders, drug-induced myopathy, ischemic heart disease and coronary artery disease [208,209,210][144][145][146].3.1. Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is one of the most common cardiac genetic conditions, with a prevalence greater than 1 in 500 in the general adult population [211][147]. This inherited disorder is characterized by left ventricular hypertrophy (>15 mm for adults), that cannot be only attributed to abnormal load conditions [212][148] (Figure 2D). Essential histopathological features include myocyte hypertrophy and disarray alongside heightened myocardial fibrosis; the combination of these hallmarks leads to left ventricular outflow track obstruction, impaired diastolic function and cardiac arrhythmias [213][149]. Genetically, HCM is an autosomal-dominant disorder caused by mutations in genes encoding for contractile and structural proteins of the cardiac muscle sarcomere apparatus [214][150]. Genetic analysis has improved our knowledge about the molecular bases of HCM, enabling clinicians to make an early identification prior to the onset of cardiac disease.3.1.1. Small Extracellular Vesicle Transcriptomic Analyses in Hypertrophic Cardiomyopathy
In this pathological scenario, James et al. (2021) [215][151] performed a transcriptomic analysis on small EVs derived from human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) with or without the c.ACTC1G301A mutation. This model of HCM [216][152] was chosen for this study as it had previously been shown to recapitulate many key disease phenotypes including abnormal contractility, Ca2+ sensitivity/handling, arrhythmogenesis and hypertrophic brain natriuretic peptide signaling. Transcriptomic analysis of HCM small EVs has shown that CMs alter their EV cargo when HCM sarcomeric mutations are present. To be more precise, they observed differences in snoRNA cargo within HCM-released small EVs that specifically is altered when HCM hiPSC-CMs were subjected to an increased workload. In total, 12 snoRNAs were identified including 10 SNORDs (SNORD6, SNOTRD116-23, SNORD116-25, SNORD116-29, SNORD18A, SNORD42A, SNORD43, SNORD58C, SNORD60, and SNORD 101) and 2 SNORAs (SNORA3B and SNORA20). The functional role of these snoRNAs is related with post-translational modifications and alternative splicing processes differentially regulated in HCM (Table 3).3.1.2. Mechanistic Insights into Small Extracellular Vesicle Related with Hypertrophic Cardiomyopathy
Some years ago, Tian et al. (2018) demonstrated an upregulation of miRNA-27a levels in both infarcted myocardial tissue and systemic circulation in a rodent model of MI. The identified miRNA-27a exhibited a propensity for incorporation into small EVs, contributing to oxidative stress and promoting hypertrophic gene expression via modulation of the Nuclear factor (erythroid-derived 2)-like 2/Kelch-like ECH-associated protein 1 (Nrf2/keap1) signaling pathway [217][153]. Moreover, clinical studies corroborated increased miRNA-27a levels in failing hearts and systemic circulation, suggesting its potential utility as a diagnostic and prognostic biomarker for HF [218,219,220][154][155][156]. Furthermore, the same lab evidenced that miR-27a* exhibited resistance to degradation and mirrored the expression pattern of miRNA-27a in chronic HF. This miR-27a* is packaged into small EVs and taken up by CM-targeting Z-line-associated protein PDLIM5, thereby contributing to hypertrophic gene expression [221][157].3.2. Dilated Cardiomyopathy
Dilated cardiomyopathy (DCM), with an incidence of 1 in 2500 individuals, is defined as the presence of left ventricular dilation along with systolic dysfunction [208,224][144][158]. Moreover, DCM is frequently associated with an increased likelihood of severe arrhythmias, which suggests the pathological affection of the cardiac conducting system. Finally, with disease progression, the right ventricle and diastolic function are affected, leading to HF and death [225][159] (Figure 2E). Some genes are associated with the initiation, progression and pathology of DCM; nonetheless, while these genes seem to be linked to DCM, only a limited number directly contribute to the onset of DCM owing to genetic variations [226,227][160][161].3.2.1. Small Extracellular Vesicle Transcriptomic Analyses in Dilated Cardiomyopathy
Zhang et al. (2023) [223][162] performed high-throughput sequencing in plasma exosomes of DCM patients with chronic heart failure (CHF) and healthy controls, and a total of 3687 miRNAs were detected in these biological samples. However, only 92 miRNAs were significantly differentially expressed between the two groups; 48 miRNAs were upregulated and 44 miRNAs were downregulated [223][162]. Six of these miRNAs have been identified as significant contributors to the development of DCM through diverse mechanisms, such as the regulation of fibrosis (miR-423-5p, hsa-miR-185-5p, hsa-miR-150-5p, hsa-miR-10a-5p_R-1) [228,229[163][164][165][166][167][168],230,231,232,233], hypertrophy (hsa-miR-150-5p) [231[166][167],232], inflammation (hsa-miR-1304-3p_1ss13CA, hsa-miR-150-5p) [231[166][167][169],232,234], oxidative stress (hsa-miR-1304-3p_1ss13CA) [234][169], angiogenesis (hsa-miR-150-5p) [235][170] and mitochondrial function (sa-miR-3138_L-5R+2) [236][171].3.2.2. Small Extracellular Vesicle Proteomic Analyses in Dilated Cardiomyopathy
Bayes-Genis laboratory explored the proteomic signature of plasma-derived small EVs obtained from DCM patients and healthy controls. A total of 176 proteins (74.6%) were shared by controls and DCM patients, whereas 51 proteins were exclusive for the DCM group and 7 proteins were exclusive for the control group [237][172]. They observed that some proteins were generally over-represented in the cargo proteome of circulating DCM small EVs compared with control small EVs. These included fibrinogen, crucially associated with a high risk of cardiovascular disease due to its contribution to endothelial injury, plasma viscosity and thrombus formation [237,238,239,240,241,242][172][173][174][175][176][177]; serotransferrin, related with anemia as comorbidity in HF patients [237,243,244,245,246,247][172][178][179][180][181][182]; protease inhibitor α-1-antitrypsin (AAT) as a putative biomarker for the evaluation of disease status [248,249][183][184]; and several apolipoproteins [237][172]. Gene ontology analysis evidenced that proteins associated with stress as well as with protein activation were found to be more abundant in DCM small EVs when compared to control samples [237][172].3.2.3. Mechanistic Insights into Small Extracellular Vesicle Related with Dilated Cardiomyopathy
Wu et al. (2018) [250][185] analyzed three different serum exosomal miRNAs, exo-miR-92b-5p, exo-miR-192-5p and exo-miR-320a, in patients with DCM and acute HF (AHF) vs. healthy volunteers. In the study, exo-miR-92b-5p was increased in DCM-AHF patients compared to control, and was finally considered as a potential biomarker that potentially predicts DCM-AHF in patients [250][185]. Recent research with angiotensin II-stimulated hiPSCs differentiated cardiomyocytes has evidenced that miR-218-5p is upregulated in the DCM-Exos. This microRNA has been identified as a critical contributor to fibrogenesis through the activation of Tgf-β signaling after the suppression of TNFAIP3 [251][186].3.2.4. Therapeutic Approaches
Several labs have reported the therapeutic role of small EVs in DCM. Vandergriff et al. (2015) [252][187] analyzed the therapeutic role of cardiac stem cell-derived exosomes (CSC-exo) in a mouse model of doxorubicin-induced DCM. Systemic delivery of human CSC-exo in mice showed improved heart function via echocardiography, as well as decreased apoptosis and fibrosis [252][187]. Sun et al. (2018) [253][188] proved that mesenchymal stem cell-derived exosomes (MSC-Exos) alleviate inflammatory cardiomyopathy by improving the inflammatory microenvironment of the myocardium, especially by regulating the activity of macrophages in a mouse model of DCM [253][188]. Ni et al. (2020) [254][189] evidenced that trophoblast stem cell-derived exosomes (TSC-exos) could alleviate DOX-induced cardiac injury via the let-7i/YAP pathway. They observed an improvement of cardiac function and decreased inflammatory responses, accompanied by downregulated YAP signaling [254][189] (Figure 2E). Zhang et al. (2022) [255][190] evidenced that small EVs derived from KLF2-overexpressing endothelial cells reduced cardiac inflammation and ameliorated left ventricular dysfunction in DCM mice by targeting the CCR2 protein to inhibit Ly6Chigh monocyte mobilization from the bone marrow [255][190].3.3. Diabetic Cardiomyopathy
The prevalence of diabetes mellitus (DM) is approximately 9.3% of the world population [256][191]. In this vast group, HF has emerged as the most common cardiovascular complication of diabetes [257][192]. Diabetic cardiomyopathy (DmCM) is a myocardial-specific complication that is associated with coronary microvascular dysfunction and increases the risk of HF in patients with diabetes [258][193] (Figure 2F). DmCM is characterized by left ventricle dysfunction, CM apoptosis and interstitial fibrosis developed in the absence of coronary artery disease, valvular disease and/or hypertension [259,260][194][195].3.3.1. Mechanistic Insights into Small Extracellular Vesicle Related with Diabetic Cardiomyopathy
Some years ago, Gonzalo-Calvo et al. (2017) [261][196] evidenced that circulating miR-1 and miR-133a levels are actively released from CM exosomes in response to lipid overload and are robustly associated with myocardial steatosis in type 2 diabetes patients [261][196]. Moreover, microRNAs which are encapsulated within exosomes offer a stable source of information to study the role of miRs associated with HF in diabetic hearts with preserved ejection fraction (HFpEF). Huang et al. (2022) [262][197] evidenced the association of exosomal miR-30d-5p and miR-126a-5p with diabetic HFpEF [262][197]. It has been shown that circulating miR-30d downregulation reduces the cardioprotective role of miR-30d in HF [263,264[198][199][200],265], whereas miR-126a downregulation decreases cardiac microvessel density and impairs ventricular function [266][201]. In addition, recent research evidenced that exosomes deliver Mst1 protein between cardiac microvascular endothelial cells (CMECs) and CM, playing a pivotal role in the development of DmCM. In this scenario, the increase in Mst1 in CM inhibits cell autophagy, enhancing the apoptotic CM ratio and, moreover, affecting glucose metabolism which leads to insulin resistance that finally contributes to DmCM and impaired cardiac function [267][202]. The same lab has recently demonstrated that exosomes mediate the interaction between CMECs and CFs. They observed that exosomes derived from CMECs under high glucose were rich in TGF-β1 mRNA, which significantly promoted the activation of CFs. This condition aggravates perivascular and interstitial fibrosis in mice with DmCM [268][203].3.3.2. Therapeutic Approaches
Heat shock protein (Hsp) response is a cellular intrinsic defense mechanism [269][204]. The expression of these proteins in type 1 and type 2 diabetes are decreased, contributing to diabetes-induced organ damage [270][205]. In this scenario, it has been evidenced that CM exosomes derived from a transgenic mouse model with cardiac-specific overexpression of Hsp20 protected against in vitro hyperglycemia-triggered cell death, as well as in vivo STZ-induced cardiac adverse remodeling. Thus, Hsp20-engineered exosomes might be a novel therapeutic agent for DmCM (Figure 2D). Moreover, Lin et al. (2019) [271][206] evidenced that mesenchymal stem cell (MSC)-derived exosomes significantly increased the levels of fatty acid transporters (FATPs) and fatty acid beta oxidase (FA-β-oxidase), whereas TGF-β1 and Smad2 mRNAs levels were significantly reduced. Such molecular regulation indicates that MSC-derived exosomes improve DM-induced myocardial injury and fibrosis via inhibition of the TGF-β1/Smad2 signaling pathway [271][206]. Similarly, parasympathetic ganglionic neuron-derived exosomes (PGN-exos) are able to inhibit apoptosis, improve cell viability and restore levels of anti-apoptotic protein Bcl-2 in diabetes-induced H9c2 cells [272][207]. Finally, recent data support the notion that ginsenoside RG1 (RG1)-induced MSCs secrete exosomes that can alleviate DmCM. Mechanistically, exosomes derived from RG1-induced MSCs transferred circNOTCH1 into macrophages, activating the NOTCH signaling pathway through the regulatory axis consisting of circNOTCH1, miR-495-3p and NOTCH1 [273][208] (Figure 2F). All these findings may contribute to the development of new therapeutic approaches for DmCM.References
- Camussi, G.; Deregibus, M.-C.; Bruno, S.; Grange, C.; Fonsato, V.; Tetta, C. Exosome/microvesicle-mediated epigenetic reprogramming of cells. Am. J. Cancer Res. 2011, 1, 98–110.
- Lee, Y.; El Andaloussi, S.; Wood, M.J.A. Exosomes and microvesicles: Extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 2012, 21, R125–R134.
- Zappulli, V.; Friis, K.P.; Fitzpatrick, Z.; Maguire, C.A.; Breakefield, X.O. Extracellular vesicles and intercellular communication within the nervous system. J. Clin. Investig. 2016, 126, 1198–1207.
- Yang, M.; Wu, S.Y. The Advances and Challenges in Utilizing Exosomes for Delivering Cancer Therapeutics. Front. Pharmacol. 2018, 9, 735.
- Witwer, K.W.; Buzás, E.I.; Bemis, L.T.; Bora, A.; Lässer, C.; Lötvall, J.; Nolte-‘t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicle 2013, 2, 20360.
- Van Der Pol, E.; Coumans, F.; Varga, Z.; Krumrey, M.; Nieuwland, R. Innovation in detection of microparticles and exosomes. J. Thromb. Haemost. 2013, 11, 36–45.
- Meldolesi, J. Exosomes and Ectosomes in Intercellular Communication. Curr. Biol. 2018, 28, R435–R444.
- Li, G.; Chen, T.; Dahlman, J.; Eniola-Adefeso, L.; Ghiran, I.C.; Kurre, P.; Lam, W.A.; Lang, J.K.; Marbán, E.; Martín, P.; et al. Current challenges and future directions for engineering extracellular vesicles for heart, lung, blood and sleep diseases. J. Extracell. Vesicle 2023, 12, 12305.
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208.
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicle 2015, 4, 27066.
- Bebelman, M.P.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11.
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383.
- Borges, F.T.; Reis, L.A.; Schor, N. Extracellular vesicles: Structure, function, and potential clinical uses in renal diseases. Braz. J. Med. Biol. Res. 2013, 46, 824–830.
- Simons, M.; Raposo, G. Exosomes—Vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581.
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593.
- Mathivanan, S.; Simpson, R.J. ExoCarta: A compendium of exosomal proteins and RNA. Proteomics 2009, 9, 4997–5000.
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727.
- Bobrie, A.; Colombo, M.; Raposo, G.; Théry, C. Exosome Secretion: Molecular Mechanisms and Roles in Immune Responses. Traffic 2011, 12, 1659–1668.
- Chaput, N.; Théry, C. Exosomes: Immune properties and potential clinical implementations. Semin. Immunopathol. 2011, 33, 419–440.
- Robbins, P.D.; Dorronsoro, A.; Booker, C.N. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J. Clin. Investig. 2016, 126, 1173–1180.
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular Vesicles: Composition, Biological Relevance, and Methods of Study. BioScience 2015, 65, 783–797.
- Rak, J. Microparticles in Cancer. Semin. Thromb. Hemost. 2010, 36, 888–906.
- Hood, J.L.; San, R.S.; Wickline, S.A. Exosomes Released by Melanoma Cells Prepare Sentinel Lymph Nodes for Tumor Metastasis. Cancer Res. 2011, 71, 3792–3801.
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289.
- Wickman, G.; Julian, L.; Olson, M.F. How apoptotic cells aid in the removal of their own cold dead bodies. Cell Death Differ. 2012, 19, 735–742.
- Théry, C.; Boussac, M.; Véron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic Analysis of Dendritic Cell-Derived Exosomes: A Secreted Subcellular Compartment Distinct from Apoptotic Vesicles. J. Immunol. 2001, 166, 7309–7318.
- Escrevente, C.; Keller, S.; Altevogt, P.; Costa, J. Interaction and uptake of exosomes by ovarian cancer cells. BMC Cancer 2011, 11, 108.
- Arraud, N.; Linares, R.; Tan, S.; Gounou, C.; Pasquet, J.-M.; Mornet, S.; Brisson, A.R. Extracellular vesicles from blood plasma: Determination of their morphology, size, phenotype and concentration. J. Thromb. Haemost. 2014, 12, 614–627.
- Sun, Y.; Xia, Z.; Shang, Z.; Sun, K.; Niu, X.; Qian, L.; Fan, L.-Y.; Cao, C.-X.; Xiao, H. Facile preparation of salivary extracellular vesicles for cancer proteomics. Sci. Rep. 2016, 6, 24669.
- Ranghino, A.; Dimuccio, V.; Papadimitriou, E.; Bussolati, B. Extracellular vesicles in the urine: Markers and mediators of tissue damage and regeneration. Clin. Kidney J. 2015, 8, 23–30.
- Théry, C. Diagnosis by extracellular vesicles. Nature 2015, 523, 161–162.
- Thompson, A.G.; Gray, E.; Heman-Ackah, S.M.; Mäger, I.; Talbot, K.; Andaloussi, S.E.; Wood, M.J.; Turner, M.R. Extracellular vesicles in neurodegenerative disease—Pathogenesis to biomarkers. Nat. Rev. Neurol. 2016, 12, 346–357.
- Turpin, D.; Truchetet, M.-E.; Faustin, B.; Augusto, J.-F.; Contin-Bordes, C.; Brisson, A.; Blanco, P.; Duffau, P. Role of extracellular vesicles in autoimmune diseases. Autoimmun. Rev. 2016, 15, 174–183.
- Loyer, X.; Vion, A.-C.; Tedgui, A.; Boulanger, C.M. Microvesicles as Cell–Cell Messengers in Cardiovascular Diseases. Circ. Res. 2014, 114, 345–353.
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; De Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics—2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603.
- WHO, Fact Sheet about Cardiovascular Diseases. 2016. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 8 December 2023).
- Gartz, M.; Strande, J.L. Examining the Paracrine Effects of Exosomes in Cardiovascular Disease and Repair. J. Am. Heart Assoc. 2018, 7, e007954.
- Chistiakov, D.; Orekhov, A.; Bobryshev, Y. Cardiac Extracellular Vesicles in Normal and Infarcted Heart. Int. J. Mol. Sci. 2016, 17, 63.
- Pironti, G.; Strachan, R.T.; Abraham, D.; Mon-Wei Yu, S.; Chen, M.; Chen, W.; Hanada, K.; Mao, L.; Watson, L.J.; Rockman, H.A. Circulating Exosomes Induced by Cardiac Pressure Overload Contain Functional Angiotensin II Type 1 Receptors. Circulation 2015, 131, 2120–2130.
- Sahoo, S.; Losordo, D.W. Exosomes and cardiac repair after myocardial infarction. Circ. Res. 2014, 114, 333–344.
- Sluijter, J.P.G.; Verhage, V.; Deddens, J.C.; Van Den Akker, F.; Doevendans, P.A. Microvesicles and exosomes for intracardiac communication. Cardiovasc. Res. 2014, 102, 302–311.
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019. J. Am. Coll. Cardiol. 2020, 76, 2982–3021.
- Saleh, M.; Ambrose, J.A. Understanding myocardial infarction. F1000Res 2018, 7, 1378.
- Jenča, D.; Melenovský, V.; Stehlik, J.; Staněk, V.; Kettner, J.; Kautzner, J.; Adámková, V.; Wohlfahrt, P. Heart failure after myocardial infarction: Incidence and predictors. ESC Heart Fail. 2021, 8, 222–237.
- Börschel, C.S.; Schnabel, R.B. The imminent epidemic of atrial fibrillation and its concomitant diseases—Myocardial infarction and heart failure—A cause for concern. Int. J. Cardiol. 2019, 287, 162–173.
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. Fourth universal definition of myocardial infarction (2018). J. Am. Coll. Cardiol. 2018, 72, 2231–2264.
- Golforoush, P.; Yellon, D.M.; Davidson, S.M. Mouse models of atherosclerosis and their suitability for the study of myocardial infarction. Basic Res. Cardiol. 2020, 115, 73.
- Scheen, A. De l’athérosclérose à l’athérothrombose. Rev. Med. Liege 2018, 73, 224–228.
- Anderson, J.L.; Morrow, D.A. Acute Myocardial Infarction. N. Engl. J. Med. 2017, 376, 2053–2064.
- Mitsis, A.; Gragnano, F. Myocardial Infarction with and without ST-segment Elevation: A Contemporary Reappraisal of Similarities and Differences. Curr. Cardiol. Rev. 2021, 17, e230421189013.
- Wang, X.; Guo, Z.; Ding, Z.; Mehta, J.L. Inflammation, Autophagy, and Apoptosis after Myocardial Infarction. J. Am. Heart Assoc. 2018, 7, e008024.
- Moreira, D.M.; Da Silva, R.L.; Vieira, J.L.; Fattah, T.; Lueneberg, M.E.; Gottschall, C.A.M. Role of Vascular Inflammation in Coronary Artery Disease: Potential of Anti-inflammatory Drugs in the Prevention of Atherothrombosis: Inflammation and Anti-Inflammatory Drugs in Coronary Artery Disease. Am. J. Cardiovasc. Drugs 2015, 15, 1–11.
- Wu, X.; Reboll, M.R.; Korf-Klingebiel, M.; Wollert, K.C. Angiogenesis after acute myocardial infarction. Cardiovasc. Res. 2021, 117, 1257–1273.
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction—From repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581.
- He, L.; Huang, X.; Kanisicak, O.; Li, Y.; Wang, Y.; Li, Y.; Pu, W.; Liu, Q.; Zhang, H.; Tian, X.; et al. Preexisting endothelial cells mediate cardiac neovascularization after injury. J. Clin. Investig. 2017, 127, 2968–2981.
- Li, Z.; Solomonidis, E.G.; Meloni, M.; Taylor, R.S.; Duffin, R.; Dobie, R.; Magalhaes, M.S.; Henderson, B.E.P.; Louwe, P.A.; D’Amico, G.; et al. Single-cell transcriptome analyses reveal novel targets modulating cardiac neovascularization by resident endothelial cells following myocardial infarction. Eur. Heart J. 2019, 40, 2507–2520.
- Loyer, X.; Zlatanova, I.; Devue, C.; Yin, M.; Howangyin, K.-Y.; Klaihmon, P.; Guerin, C.L.; Kheloufi, M.; Vilar, J.; Zannis, K.; et al. Intra-Cardiac Release of Extracellular Vesicles Shapes Inflammation Following Myocardial Infarction. Circ. Res. 2018, 123, 100–106.
- Pan, W.; Zhu, Y.; Meng, X.; Zhang, C.; Yang, Y.; Bei, Y. Immunomodulation by Exosomes in Myocardial Infarction. J. Cardiovasc. Trans. Res. 2019, 12, 28–36.
- Xu, Y.; Wu, A.; Chen, J.; Song, X.; Chen, M.; Liu, Q. Limb-Bud and Heart (LBH) Upregulation in Cardiomyocytes under Hypoxia Promotes the Activation of Cardiac Fibroblasts via Exosome Secretion. Mediat. Inflamm. 2022, 2022, 8939449.
- Nian, W.; Fu, C. Exosomes in Myocardial Infarction: Therapeutic Potential and Clinical Application. J. Cardiovasc. Trans. Res. 2023, 16, 87–96.
- Gong, Z.; Wen, M.; Zhang, W.; Yu, L.; Huang, C.; Xu, Y.; Xia, Z.; Xu, M.; Xu, J.; Liang, Q.; et al. Plasma exosomes induce inflammatory immune response in patients with acute myocardial infarction. Arch. Physiol. Biochem. 2023, 129, 1168–1176.
- Zhang, Z.; Xu, Y.; Cao, C.; Wang, B.; Guo, J.; Qin, Z.; Lu, Y.; Zhang, J.; Zhang, L.; Wang, W.; et al. Exosomes as a messager to regulate the crosstalk between macrophages and cardiomyocytes under hypoxia conditions. J. Cell. Mol. Medi 2022, 26, 1486–1500.
- Cheng, M.; An, S.; Li, J. Identifying key genes associated with acute myocardial infarction. Medicine 2017, 96, e7741.
- Keller, T.; Zeller, T.; Peetz, D.; Tzikas, S.; Roth, A.; Czyz, E.; Bickel, C.; Baldus, S.; Warnholtz, A.; Fröhlich, M.; et al. Sensitive Troponin I Assay in Early Diagnosis of Acute Myocardial Infarction. N. Engl. J. Med. 2009, 361, 868–877.
- Ai, J.; Zhang, R.; Li, Y.; Pu, J.; Lu, Y.; Jiao, J.; Li, K.; Yu, B.; Li, Z.; Wang, R.; et al. Circulating microRNA-1 as a potential novel biomarker for acute myocardial infarction. Biochem. Biophys. Res. Commun. 2010, 391, 73–77.
- Wang, G.-K.; Zhu, J.-Q.; Zhang, J.-T.; Li, Q.; Li, Y.; He, J.; Qin, Y.-W.; Jing, Q. Circulating microRNA: A novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur. Heart J. 2010, 31, 659–666.
- Liu, P.; Wang, S.; Li, K.; Yang, Y.; Man, Y.; Du, F.; Wang, L.; Tian, J.; Su, G. Exosomal microRNA-4516, microRNA-203 and SFRP1 are potential biomarkers of acute myocardial infarction. Mol. Med. Rep. 2023, 27, 124.
- Chen, S.; Fang, H.; Liu, R.; Fang, Y.; Wu, Z.; Xie, P. miR-6718-5p and miR-4329 can be used as potential biomarkers for acute myocardial infarction. J. Card. Surg. 2021, 36, 3721–3728.
- Guo, M.; Li, R.; Yang, L.; Zhu, Q.; Han, M.; Chen, Z.; Ruan, F.; Yuan, Y.; Liu, Z.; Huang, B.; et al. Evaluation of exosomal miRNAs as potential diagnostic biomarkers for acute myocardial infarction using next-generation sequencing. Ann. Transl. Med. 2021, 9, 219.
- Zhao, X.; Jia, Y.; Chen, H.; Yao, H.; Guo, W. Plasma-derived exosomal miR-183 associates with protein kinase activity and may serve as a novel predictive biomarker of myocardial ischemic injury. Exp. Ther. Med. 2019, 18, 179–187.
- Ling, H.; Guo, Z.; Shi, Y.; Zhang, L.; Song, C. Serum Exosomal MicroRNA-21, MicroRNA-126, and PTEN Are Novel Biomarkers for Diagnosis of Acute Coronary Syndrome. Front. Physiol. 2020, 11, 654.
- Hu, J.; Huang, C.-X.; Rao, P.-P.; Zhou, J.-P.; Wang, X.; Tang, L.; Liu, M.-X.; Zhang, G.-G. Inhibition of microRNA-155 attenuates sympathetic neural remodeling following myocardial infarction via reducing M1 macrophage polarization and inflammatory responses in mice. Eur. J. Pharmacol. 2019, 851, 122–132.
- Hu, C.; Liao, J.; Huang, R.; Su, Q.; He, L. MicroRNA-155-5p in serum derived-exosomes promotes ischaemia–reperfusion injury by reducing CypD ubiquitination by NEDD4. ESC Heart Fail. 2023, 10, 1144–1157.
- Chen, C.; Cai, S.; Wu, M.; Wang, R.; Liu, M.; Cao, G.; Dong, M.; Yiu, K.-H. Role of Cardiomyocyte-Derived Exosomal MicroRNA-146a-5p in Macrophage Polarization and Activation. Dis. Markers 2022, 2022, 2948578.
- Geng, T.; Song, Z.-Y.; Xing, J.-X.; Wang, B.-X.; Dai, S.-P.; Xu, Z.-S. Exosome Derived from Coronary Serum of Patients with Myocardial Infarction Promotes Angiogenesis through the miRNA-143/IGF-IR Pathway. Int. J. Nanomed. 2020, 15, 2647–2658.
- Ribeiro-Rodrigues, T.M.; Laundos, T.L.; Pereira-Carvalho, R.; Batista-Almeida, D.; Pereira, R.; Coelho-Santos, V.; Silva, A.P.; Fernandes, R.; Zuzarte, M.; Enguita, F.J.; et al. Exosomes secreted by cardiomyocytes subjected to ischaemia promote cardiac angiogenesis. Cardiovasc. Res. 2017, 113, 1338–1350.
- Liu, H.; Zhang, Y.; Yuan, J.; Gao, W.; Zhong, X.; Yao, K.; Lin, L.; Ge, J. Dendritic cell-derived exosomal miR-494-3p promotes angiogenesis following myocardial infarction. Int. J. Mol. Med. 2020, 47, 315–325.
- Li, H.; Ding, J.; Liu, W.; Wang, X.; Feng, Y.; Guan, H.; Chen, Z. Plasma exosomes from patients with acute myocardial infarction alleviate myocardial injury by inhibiting ferroptosis through miR-26b-5p/SLC7A11 axis. Life Sci. 2023, 322, 121649.
- Sun, P.; Wang, C.; Mang, G.; Xu, X.; Fu, S.; Chen, J.; Wang, X.; Wang, W.; Li, H.; Zhao, P.; et al. Extracellular vesicle-packaged mitochondrial disturbing miRNA exacerbates cardiac injury during acute myocardial infarction. Clin. Transl. Med. 2022, 12, e779.
- Zheng, M.; Liu, X.; Han, R.; Yuan, W.; Sun, K.; Zhong, J.; Yang, X. Circulating exosomal long non-coding RNAs in patients with acute myocardial infarction. J. Cell. Mol. Med. 2020, 24, 9388–9396.
- Dang, Y.; Hua, W.; Zhang, X.; Sun, H.; Zhang, Y.; Yu, B.; Wang, S.; Zhang, M.; Kong, Z.; Pan, D.; et al. Anti-angiogenic effect of exo-LncRNA TUG1 in myocardial infarction and modulation by remote ischemic conditioning. Basic Res. Cardiol. 2023, 118, 1.
- Lin, B.; Chen, X.; Lu, C.; Xu, J.; Qiu, Y.; Liu, X.; Song, H.; Chen, A.; Xiong, J.; Wang, K.; et al. Loss of exosomal LncRNA HCG15 prevents acute myocardial ischemic injury through the NF-κB/p65 and p38 pathways. Cell Death Dis. 2021, 12, 1007.
- Wang, W.; Wang, Y.; Piao, H.; Li, B.; Huang, M.; Zhu, Z.; Li, D.; Wang, T.; Xu, R.; Liu, K. Circular RNAs as potential biomarkers and therapeutics for cardiovascular disease. PeerJ 2019, 7, e6831.
- Wang, G.; Wang, C.; Huang, Z.; Sun, S.; Chen, Y. Exosomal circ-0020887 and circ-0009590 as novel biomarkers for the diagnosis and prediction of short-term adverse cardiovascular outcomes in STEMI patients. Open Med. 2023, 18, 20230807.
- Zhu, J.; Chen, Z.; Peng, X.; Zheng, Z.; Le, A.; Guo, J.; Ma, L.; Shi, H.; Yao, K.; Zhang, S.; et al. Extracellular Vesicle-Derived circITGB1 Regulates Dendritic Cell Maturation and Cardiac Inflammation via miR-342-3p/NFAM1. Oxidative Med. Cell. Longev. 2022, 2022, 8392313.
- Wang, Y.; Zhao, R.; Liu, W.; Wang, Z.; Rong, J.; Long, X.; Liu, Z.; Ge, J.; Shi, B. Exosomal circHIPK3 Released from Hypoxia-Pretreated Cardiomyocytes Regulates Oxidative Damage in Cardiac Microvascular Endothelial Cells via the miR-29a/IGF-1 Pathway. Oxidative Med. Cell. Longev. 2019, 2019, 7954657.
- Lan, Z.; Wang, T.; Zhang, L.; Jiang, Z.; Zou, X. CircSLC8A1 Exacerbates Hypoxia-Induced Myocardial Injury via Interacting with MiR-214-5p to Upregulate TEAD1 Expression. Int. Heart J. 2022, 63, 591–601.
- Wang, Y.; Zhao, R.; Shen, C.; Liu, W.; Yuan, J.; Li, C.; Deng, W.; Wang, Z.; Zhang, W.; Ge, J.; et al. Exosomal CircHIPK3 Released from Hypoxia-Induced Cardiomyocytes Regulates Cardiac Angiogenesis after Myocardial Infarction. Oxidative Med. Cell. Longev. 2020, 2020, 8418407.
- Fan, M.; Liang, T.; Xie, F.; Ma, P.; Li, J. Exosomal circ_HIPK3 reduces apoptosis in H2O2-induced AC16 cardiomyocytes through miR-33a-5p/IRS1 axis. Transpl. Immunol. 2023, 80, 101862.
- Xie, Y.; Zhang, H.; Huang, T. Quantitative proteomics reveal three potential biomarkers for risk assessment of acute myocardial infarction. Bioengineered 2022, 13, 4939–4950.
- Chen, Z.; Yan, Y.; Wu, J.; Qi, C.; Liu, J.; Wang, J. Expression level and diagnostic value of exosomal NEAT1/miR-204/MMP-9 in acute ST-segment elevation myocardial infarction. IUBMB Life 2020, 72, 2499–2507.
- Foglio, E.; Puddighinu, G.; Fasanaro, P.; D’Arcangelo, D.; Perrone, G.A.; Mocini, D.; Campanella, C.; Coppola, L.; Logozzi, M.; Azzarito, T.; et al. Exosomal clusterin, identified in the pericardial fluid, improves myocardial performance following MI through epicardial activation, enhanced arteriogenesis and reduced apoptosis. Int. J. Cardiol. 2015, 197, 333–347.
- Li, Z.; Huo, X.; Chen, K.; Yang, F.; Tan, W.; Zhang, Q.; Yu, H.; Li, C.; Zhou, D.; Chen, H.; et al. Profilin 2 and Endothelial Exosomal Profilin 2 Promote Angiogenesis and Myocardial Infarction Repair in Mice. Front. Cardiovasc. Med. 2022, 9, 781753.
- Gidlöf, O.; Evander, M.; Rezeli, M.; Marko-Varga, G.; Laurell, T.; Erlinge, D. Proteomic profiling of extracellular vesicles reveals additional diagnostic biomarkers for myocardial infarction compared to plasma alone. Sci. Rep. 2019, 9, 8991.
- Wang, C.; Zhang, C.; Liu, L.; Xi, A.; Chen, B.; Li, Y.; Du, J. Macrophage-Derived mir-155-Containing Exosomes Suppress Fibroblast Proliferation and Promote Fibroblast Inflammation during Cardiac Injury. Mol. Ther. 2017, 25, 192–204.
- Liu, S.; Chen, J.; Shi, J.; Zhou, W.; Wang, L.; Fang, W.; Zhong, Y.; Chen, X.; Chen, Y.; Sabri, A.; et al. M1-like macrophage-derived exosomes suppress angiogenesis and exacerbate cardiac dysfunction in a myocardial infarction microenvironment. Basic Res. Cardiol. 2020, 115, 22.
- Cai, L.; Chao, G.; Li, W.; Zhu, J.; Li, F.; Qi, B.; Wei, Y.; Chen, S.; Zhou, G.; Lu, X.; et al. Activated CD4+ T cells-derived exosomal miR-142-3p boosts post-ischemic ventricular remodeling by activating myofibroblast. Aging 2020, 12, 7380–7396.
- Gou, L.; Xue, C.; Tang, X.; Fang, Z. Inhibition of Exo-miR-19a-3p derived from cardiomyocytes promotes angiogenesis and improves heart function in mice with myocardial infarction via targeting HIF-1α. Aging 2020, 12, 23609–23618.
- Huang, J.; Wang, F.; Sun, X.; Chu, X.; Jiang, R.; Wang, Y.; Pang, L. Myocardial infarction cardiomyocytes-derived exosomal miR-328-3p promote apoptosis via Caspase signaling. Am. J. Transl. Res. 2021, 13, 2365–2378.
- Yang, J.; Yu, X.; Xue, F.; Li, Y.; Liu, W.; Zhang, S. Exosomes derived from cardiomyocytes promote cardiac fibrosis via myocyte-fibroblast cross-talk. Am. J. Transl. Res. 2018, 10, 4350–4366.
- Jiang, W.; Song, Q.; Lu, Z.; Wang, S.; Liu, T.; Wang, X.; Wang, M. Myocardial Infarction-Associated Extracellular Vesicle-Delivered miR-208b Affects the Growth of Human Umbilical Vein Endothelial Cells via Regulating CDKN1A. BioMed Res. Int. 2021, 2021, 9965639.
- Wang, X.; Morelli, M.B.; Matarese, A.; Sardu, C.; Santulli, G. Cardiomyocyte-derived exosomal microRNA-92a mediates post-ischemic myofibroblast activation both in vitro and ex vivo. ESC Heart Fail. 2020, 7, 285–289.
- Yang, Y.; Li, Y.; Chen, X.; Cheng, X.; Liao, Y.; Yu, X. Exosomal transfer of miR-30a between cardiomyocytes regulates autophagy after hypoxia. J. Mol. Med. 2016, 94, 711–724.
- Wang, B.; Cao, C.; Han, D.; Bai, J.; Guo, J.; Guo, Q.; Li, D.; Zhang, J.; Zhang, Z.; Wang, Y.; et al. Dysregulation of miR-342-3p in plasma exosomes derived from convalescent AMI patients and its consequences on cardiac repair. Biomed. Pharmacother. 2021, 142, 112056.
- Sun, S.; Wu, Y.; Maimaitijiang, A.; Huang, Q.; Chen, Q. Ferroptotic cardiomyocyte-derived exosomes promote cardiac macrophage M1 polarization during myocardial infarction. PeerJ 2022, 10, e13717.
- Zhang, Q.; Chen, L.; Huang, L.; Cheng, H.; Wang, L.; Xu, L.; Hu, D.; He, C.; Fu, C.; Wei, Q. CD44 promotes angiogenesis in myocardial infarction through regulating plasma exosome uptake and further enhancing FGFR2 signaling transduction. Mol. Med. 2022, 28, 145.
- Zhang, Y.; Gao, W.; Yuan, J.; Zhong, X.; Yao, K.; Luo, R.; Liu, H. CCR7 Mediates Dendritic-Cell-Derived Exosome Migration and Improves Cardiac Function after Myocardial Infarction. Pharmaceutics 2023, 15, 461.
- Liu, H.; Gao, W.; Yuan, J.; Wu, C.; Yao, K.; Zhang, L.; Ma, L.; Zhu, J.; Zou, Y.; Ge, J. Exosomes derived from dendritic cells improve cardiac function via activation of CD4+ T lymphocytes after myocardial infarction. J. Mol. Cell. Cardiol. 2016, 91, 123–133.
- Yue, Y.; Garikipati, V.N.S.; Verma, S.K.; Goukassian, D.A.; Kishore, R. Interleukin-10 Deficiency Impairs Reparative Properties of Bone Marrow-Derived Endothelial Progenitor Cell Exosomes. Tissue Eng. Part A 2017, 23, 1241–1250.
- Yue, Y.; Wang, C.; Benedict, C.; Huang, G.; Truongcao, M.; Roy, R.; Cimini, M.; Garikipati, V.N.S.; Cheng, Z.; Koch, W.J.; et al. Interleukin-10 Deficiency Alters Endothelial Progenitor Cell–Derived Exosome Reparative Effect on Myocardial Repair via Integrin-Linked Kinase Enrichment. Circ. Res. 2020, 126, 315–329.
- Zhang, Y.-Q.; Hong, L.; Jiang, Y.-F.; Hu, S.-D.; Zhang, N.-N.; Xu, L.-B.; Li, H.-X.; Xu, G.-D.; Zhou, Y.-F.; Sun, K.-Y. hAECs and their exosomes improve cardiac function after acute myocardial infarction in rats. Aging 2021, 13, 15032–15043.
- Deng, S.; Zhou, X.; Ge, Z.; Song, Y.; Wang, H.; Liu, X.; Zhang, D. Exosomes from adipose-derived mesenchymal stem cells ameliorate cardiac damage after myocardial infarction by activating S1P/SK1/S1PR1 signaling and promoting macrophage M2 polarization. Int. J. Biochem. Cell Biol. 2019, 114, 105564.
- Hu, H.; Wu, J.; Cao, C.; Ma, L. Exosomes derived from regulatory T cells ameliorate acute myocardial infarction by promoting macrophage M2 polarization. IUBMB Life 2020, 72, 2409–2419.
- Liu, L.; Jin, X.; Hu, C.-F.; Li, R.; Zhou, Z.; Shen, C.-X. Exosomes Derived from Mesenchymal Stem Cells Rescue Myocardial Ischaemia/Reperfusion Injury by Inducing Cardiomyocyte Autophagy Via AMPK and Akt Pathways. Cell Physiol. Biochem. 2017, 43, 52–68.
- Zhu, D.; Liu, S.; Huang, K.; Wang, Z.; Hu, S.; Li, J.; Li, Z.; Cheng, K. Intrapericardial Exosome Therapy Dampens Cardiac Injury via Activating Foxo3. Circ. Res. 2022, 131, e135–e150.
- Jiao, W.; Hao, J.; Xie, Y.; Meng, M.; Gao, W. EZH2 mitigates the cardioprotective effects of mesenchymal stem cell-secreted exosomes against infarction via HMGA2-mediated PI3K/AKT signaling. BMC Cardiovasc. Disord. 2022, 22, 95.
- Xu, R.; Zhang, F.; Chai, R.; Zhou, W.; Hu, M.; Liu, B.; Chen, X.; Liu, M.; Xu, Q.; Liu, N.; et al. Exosomes derived from pro-inflammatory bone marrow-derived mesenchymal stem cells reduce inflammation and myocardial injury via mediating macrophage polarization. J. Cell. Mol. Med. 2019, 23, 7617–7631.
- Liao, Z.; Chen, Y.; Duan, C.; Zhu, K.; Huang, R.; Zhao, H.; Hintze, M.; Pu, Q.; Yuan, Z.; Lv, L.; et al. Cardiac telocytes inhibit cardiac microvascular endothelial cell apoptosis through exosomal miRNA-21-5p-targeted cdip1 silencing to improve angiogenesis following myocardial infarction. Theranostics 2021, 11, 268–291.
- Gu, H.; Liu, Z.; Li, Y.; Xie, Y.; Yao, J.; Zhu, Y.; Xu, J.; Dai, Q.; Zhong, C.; Zhu, H.; et al. Serum-Derived Extracellular Vesicles Protect Against Acute Myocardial Infarction by Regulating miR-21/PDCD4 Signaling Pathway. Front. Physiol. 2018, 9, 348.
- Wei, Z.; Chen, Z.; Zhao, Y.; Fan, F.; Xiong, W.; Song, S.; Yin, Y.; Hu, J.; Yang, K.; Yang, L.; et al. Mononuclear phagocyte system blockade using extracellular vesicles modified with CD47 on membrane surface for myocardial infarction reperfusion injury treatment. Biomaterials 2021, 275, 121000.
- Zhang, J.; Lu, Y.; Mao, Y.; Yu, Y.; Wu, T.; Zhao, W.; Zhu, Y.; Zhao, P.; Zhang, F. IFN-γ enhances the efficacy of mesenchymal stromal cell-derived exosomes via miR-21 in myocardial infarction rats. Stem. Cell Res. Ther. 2022, 13, 333.
- Luther, K.M.; Haar, L.; McGuinness, M.; Wang, Y.; Lynch Iv, T.L.; Phan, A.; Song, Y.; Shen, Z.; Gardner, G.; Kuffel, G.; et al. Exosomal miR-21a-5p mediates cardioprotection by mesenchymal stem cells. J. Mol. Cell. Cardiol. 2018, 119, 125–137.
- Zhu, L.-P.; Tian, T.; Wang, J.-Y.; He, J.-N.; Chen, T.; Pan, M.; Xu, L.; Zhang, H.; Qiu, X.-T.; Li, C.-C.; et al. Hypoxia-elicited mesenchymal stem cell-derived exosomes facilitates cardiac repair through miR-125b-mediated prevention of cell death in myocardial infarction. Theranostics 2018, 8, 6163–6177.
- Xiao, C.; Wang, K.; Xu, Y.; Hu, H.; Zhang, N.; Wang, Y.; Zhong, Z.; Zhao, J.; Li, Q.; Zhu, D.; et al. Transplanted Mesenchymal Stem Cells Reduce Autophagic Flux in Infarcted Hearts via the Exosomal Transfer of miR-125b. Circ. Res. 2018, 123, 564–578.
- Ma, T.; Chen, Y.; Chen, Y.; Meng, Q.; Sun, J.; Shao, L.; Yu, Y.; Huang, H.; Hu, Y.; Yang, Z.; et al. MicroRNA-132, Delivered by Mesenchymal Stem Cell-Derived Exosomes, Promote Angiogenesis in Myocardial Infarction. Stem. Cells Int. 2018, 2018, 3290372.
- Wang, X.; Zhao, Y.; Sun, L.; Shi, Y.; Li, Z.; Zhao, X.; Xu, C.; Ji, H.; Wang, M.; Xu, W.; et al. Exosomes derived from human umbilical cord mesenchymal stem cells improve myocardial repair via upregulation of Smad7. Int. J. Mol. Med. 2018, 41, 3063–3072.
- Yang, M.; Liao, M.; Liu, R.; Zhang, Q.; Zhang, S.; He, Y.; Jin, J.; Zhang, P.; Zhou, L. Human umbilical cord mesenchymal stem cell-derived extracellular vesicles loaded with miR-223 ameliorate myocardial infarction through P53/S100A9 axis. Genomics 2022, 114, 110319.
- Song, Y.; Wang, B.; Zhu, X.; Hu, J.; Sun, J.; Xuan, J.; Ge, Z. Human umbilical cord blood–derived MSCs exosome attenuate myocardial injury by inhibiting ferroptosis in acute myocardial infarction mice. Cell Biol. Toxicol. 2021, 37, 51–64.
- Zhu, W.; Sun, L.; Zhao, P.; Liu, Y.; Zhang, J.; Zhang, Y.; Hong, Y.; Zhu, Y.; Lu, Y.; Zhao, W.; et al. Macrophage migration inhibitory factor facilitates the therapeutic efficacy of mesenchymal stem cells derived exosomes in acute myocardial infarction through upregulating miR-133a-3p. J. Nanobiotechnol. 2021, 19, 61.
- De Couto, G.; Gallet, R.; Cambier, L.; Jaghatspanyan, E.; Makkar, N.; Dawkins, J.F.; Berman, B.P.; Marbán, E. Exosomal MicroRNA Transfer Into Macrophages Mediates Cellular Postconditioning. Circulation 2017, 136, 200–214.
- Wei, Z.; Qiao, S.; Zhao, J.; Liu, Y.; Li, Q.; Wei, Z.; Dai, Q.; Kang, L.; Xu, B. miRNA-181a over-expression in mesenchymal stem cell-derived exosomes influenced inflammatory response after myocardial ischemia-reperfusion injury. Life Sci. 2019, 232, 116632.
- Li, X.; Hu, X.; Chen, Q.; Jiang, T. Bone marrow mesenchymal stem cell-derived exosomes carrying E3 ubiquitin ligase ITCH attenuated cardiomyocyte apoptosis by mediating apoptosis signal-regulated kinase-1. Pharmacogenet. Genom. 2023, 33, 117–125.
- Ning, H.; Chen, H.; Deng, J.; Xiao, C.; Xu, M.; Shan, L.; Yang, C.; Zhang, Z. Exosomes secreted by FNDC5-BMMSCs protect myocardial infarction by anti-inflammation and macrophage polarization via NF-κB signaling pathway and Nrf2/HO-1 axis. Stem Cell Res. Ther. 2021, 12, 519.
- Gong, X.; Liu, H.; Wang, S.; Liang, S.; Wang, G. Exosomes derived from SDF1-overexpressing mesenchymal stem cells inhibit ischemic myocardial cell apoptosis and promote cardiac endothelial microvascular regeneration in mice with myocardial infarction. J. Cell. Physiol. 2019, 234, 13878–13893.
- Xuan, W.; Khan, M.; Ashraf, M. Extracellular Vesicles From Notch Activated Cardiac Mesenchymal Stem Cells Promote Myocyte Proliferation and Neovasculogenesis. Front. Cell Dev. Biol. 2020, 8, 11.
- Liu, D.; Gu, G.; Gan, L.; Yan, W.; Zhang, Z.; Yao, P.; Zhu, D.; Lau, W.B.; Xie, D.; Wu, S.; et al. Identification of a CTRP9 C-Terminal polypeptide capable of enhancing bone-derived mesenchymal stem cell cardioprotection through promoting angiogenic exosome production. Redox Biol. 2021, 41, 101929.
- Liu, X.; Li, X.; Zhu, W.; Zhang, Y.; Hong, Y.; Liang, X.; Fan, B.; Zhao, H.; He, H.; Zhang, F. Exosomes from mesenchymal stem cells overexpressing MIF enhance myocardial repair. J. Cell. Physiol. 2020, 235, 8010–8022.
- Wang, Q.; Zhang, L.; Sun, Z.; Chi, B.; Zou, A.; Mao, L.; Xiong, X.; Jiang, J.; Sun, L.; Zhu, W.; et al. HIF-1α overexpression in mesenchymal stem cell-derived exosome-encapsulated arginine-glycine-aspartate (RGD) hydrogels boost therapeutic efficacy of cardiac repair after myocardial infarction. Mater. Today Bio 2021, 12, 100171.
- Sun, J.; Shen, H.; Shao, L.; Teng, X.; Chen, Y.; Liu, X.; Yang, Z.; Shen, Z. HIF-1α overexpression in mesenchymal stem cell-derived exosomes mediates cardioprotection in myocardial infarction by enhanced angiogenesis. Stem Cell Res. Ther. 2020, 11, 373.
- He, J.-G.; Li, H.-R.; Han, J.-X.; Li, B.-B.; Yan, D.; Li, H.-Y.; Wang, P.; Luo, Y. GATA-4-expressing mouse bone marrow mesenchymal stem cells improve cardiac function after myocardial infarction via secreted exosomes. Sci. Rep. 2018, 8, 9047.
- Hao, C.; Lu, Z.; Zhao, Y.; Chen, Z.; Shen, C.; Ma, G.; Chen, L. Overexpression of GATA4 enhances the antiapoptotic effect of exosomes secreted from cardiac colony-forming unit fibroblasts via miRNA221-mediated targeting of the PTEN/PI3K/AKT signaling pathway. Stem Cell Res. Ther. 2020, 11, 251.
- Yu, B.; Kim, H.W.; Gong, M.; Wang, J.; Millard, R.W.; Wang, Y.; Ashraf, M.; Xu, M. Exosomes secreted from GATA-4 overexpressing mesenchymal stem cells serve as a reservoir of anti-apoptotic microRNAs for cardioprotection. Int. J. Cardiol. 2015, 182, 349–360.
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur. Heart J. 2007, 29, 270–276.
- Richardson, P.; McKenna, W.; Bristow, M.; Maisch, B.; Mautner, B.; O’Connell, J.; Olsen, E.; Thiene, G.; Goodwin, J.; Gyarfas, I.; et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of Cardiomyopathies. Circulation 1996, 93, 841–842.
- Jenni, R. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: A step towards classification as a distinct cardiomyopathy. Heart 2001, 86, 666–671.
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B. Contemporary Definitions and Classification of the Cardiomyopathies: An American Heart Association Scientific Statement From the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006, 113, 1807–1816.
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New Perspectives on the Prevalence of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254.
- Medical Masterclass contributors; Firth, J. Cardiology: Hypertrophic cardiomyopathy. Clin. Med. 2019, 19, 61–63.
- Halliday, B.P.; Prasad, S.K. The Interstitium in the Hypertrophied Heart. JACC Cardiovasc. Imaging 2019, 12, 2357–2368.
- Maron, B.J.; Maron, M.S. Hypertrophic cardiomyopathy. Lancet 2013, 381, 242–255.
- James, V.; Nizamudeen, Z.A.; Lea, D.; Dottorini, T.; Holmes, T.L.; Johnson, B.B.; Arkill, K.P.; Denning, C.; Smith, J.G.W. Transcriptomic Analysis of Cardiomyocyte Extracellular Vesicles in Hypertrophic Cardiomyopathy Reveals Differential snoRNA Cargo. Stem Cells Dev. 2021, 30, 1215–1227.
- Smith, J.G.W.; Owen, T.; Bhagwan, J.R.; Mosqueira, D.; Scott, E.; Mannhardt, I.; Patel, A.; Barriales-Villa, R.; Monserrat, L.; Hansen, A.; et al. Isogenic Pairs of hiPSC-CMs with Hypertrophic Cardiomyopathy/LVNC-Associated ACTC1 E99K Mutation Unveil Differential Functional Deficits. Stem Cell Rep. 2018, 11, 1226–1243.
- Tian, C.; Gao, L.; Zimmerman, M.C.; Zucker, I.H. Myocardial infarction-induced microRNA-enriched exosomes contribute to cardiac Nrf2 dysregulation in chronic heart failure. Am. J. Physiol.-Heart Circ. Physiol. 2018, 314, H928–H939.
- Matkovich, S.J.; Van Booven, D.J.; Youker, K.A.; Torre-Amione, G.; Diwan, A.; Eschenbacher, W.H.; Dorn, L.E.; Watson, M.A.; Margulies, K.B.; Dorn, G.W. Reciprocal Regulation of Myocardial microRNAs and Messenger RNA in Human Cardiomyopathy and Reversal of the microRNA Signature by Biomechanical Support. Circulation 2009, 119, 1263–1271.
- Kuosmanen, S.M.; Hartikainen, J.; Hippeläinen, M.; Kokki, H.; Levonen, A.-L.; Tavi, P. MicroRNA Profiling of Pericardial Fluid Samples from Patients with Heart Failure. PLoS ONE 2015, 10, e0119646.
- Ovchinnikova, E.S.; Schmitter, D.; Vegter, E.L.; Ter Maaten, J.M.; Valente, M.A.E.; Liu, L.C.Y.; Van Der Harst, P.; Pinto, Y.M.; De Boer, R.A.; Meyer, S.; et al. Signature of circulating microRNAs in patients with acute heart failure. Eur. J. Heart Fail. 2016, 18, 414–423.
- Tian, C.; Hu, G.; Gao, L.; Hackfort, B.T.; Zucker, I.H. Extracellular vesicular MicroRNA-27a* contributes to cardiac hypertrophy in chronic heart failure. J. Mol. Cell. Cardiol. 2020, 143, 120–131.
- Cohn, J.N.; Bristow, M.R.; Chien, K.R.; Colucci, W.S.; Frazier, O.H.; Leinwand, L.A.; Lorell, B.H.; Moss, A.J.; Sonnenblick, E.H.; Walsh, R.A.; et al. Report of the National Heart, Lung, and Blood Institute Special Emphasis Panel on Heart Failure Research. Circulation 1997, 95, 766–770.
- Masarone, D.; Kaski, J.P.; Pacileo, G.; Elliott, P.M.; Bossone, E.; Day, S.M.; Limongelli, G. Epidemiology and Clinical Aspects of Genetic Cardiomyopathies. Heart Fail. Clin. 2018, 14, 119–128.
- Sweet, M.E.; Taylor, M.R.; Mestroni, L. Diagnosis, prevalence, and screening of familial dilated cardiomyopathy. Expert Opin. Orphan Drugs 2015, 3, 869–876.
- Harakalova, M.; Kummeling, G.; Sammani, A.; Linschoten, M.; Baas, A.F.; Van Der Smagt, J.; Doevendans, P.A.; Van Tintelen, J.P.; Dooijes, D.; Mokry, M.; et al. A systematic analysis of genetic dilated cardiomyopathy reveals numerous ubiquitously expressed and muscle-specific genes. Eur. J. Heart Fail. 2015, 17, 484–493.
- Zhang, L.; Zhang, G.; Lu, Y.; Gao, J.; Qin, Z.; Xu, S.; Wang, Z.; Xu, Y.; Yang, Y.; Zhang, J.; et al. Differential expression profiles of plasma exosomal microRNAs in dilated cardiomyopathy with chronic heart failure. J. Cell. Mol. Med. 2023, 27, 1988–2003.
- Bursa, F.; Yellowlees, A.; Bishop, A.; Beckett, A.; Hallis, B.; Matheson, M. Estimation of ELISA results using a parallel curve analysis. J. Immunol. Methods 2020, 486, 112836.
- Lin, R.; Rahtu-Korpela, L.; Szabo, Z.; Kemppi, A.; Skarp, S.; Kiviniemi, A.M.; Lepojärvi, E.S.; Halmetoja, E.; Kilpiö, T.; Porvari, K.; et al. MiR-185-5p regulates the development of myocardial fibrosis. J. Mol. Cell. Cardiol. 2022, 165, 130–140.
- Saincher, R.; Pentapati, K.; Gadicherla, S. Effect of audio-visual treatment information on hemodynamic parameters during the transalveolar extraction of mandibular third molars: A randomized clinical trial. J. Int. Soc. Prev. Communit. Dent. 2019, 9, 21.
- Zahid, M.; Rinas, U. Guidelines for Small-Scale Production and Purification of Hepatitis B Surface Antigen Virus-Like Particles from Recombinant Pichia pastoris. In Recombinant Protein Production in Yeast; Gasser, B., Mattanovich, D., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1923, pp. 309–322. ISBN 978-1-4939-9023-8.
- Sigterman, T.A.; Bolt, L.J.J.; Snoeijs, M.G.; Krasznai, A.G.; Heijboer, R.; Schurink, G.-W.H.; Bouwman, L.H. Radiation Exposure during Percutaneous Transluminal Angioplasty for Symptomatic Peripheral Arterial Disease. Ann. Vasc. Surg. 2016, 33, 167–172.
- Linardi, D.; Walpoth, B.; Mani, R.; Murari, A.; Tessari, M.; Hoxha, S.; Anderloni, M.; Decimo, I.; Dolci, S.; Nicolato, E.; et al. Slow versus fast rewarming after hypothermic circulatory arrest: Effects on neuroinflammation and cerebral oedema. Eur. J. Cardio-Thorac. Surg. 2020, 58, 792–800.
- Muranishi, Y.; Tanaka, N.; Kono, T.; Miyahara, R. A case of intrapulmonary lymphangioma surrounded by pulmonary hilar structures. Respir. Investig. 2020, 58, 506–509.
- Dover, E.N.; Beck, R.; Huang, M.C.; Douillet, C.; Wang, Z.; Klett, E.L.; Stýblo, M. Arsenite and methylarsonite inhibit mitochondrial metabolism and glucose-stimulated insulin secretion in INS-1 832/13 β cells. Arch. Toxicol. 2018, 92, 693–704.
- Kohanski, M.A.; Palmer, J.N.; Adappa, N.D. Indications and endonasal treatment of petrous apex cholesterol granulomas. Curr. Opin. Otolaryngol. Head Neck Surg. 2019, 27, 54–58.
- Roura, S.; Gámez-Valero, A.; Lupón, J.; Gálvez-Montón, C.; Borràs, F.E.; Bayes-Genis, A. Proteomic signature of circulating extracellular vesicles in dilated cardiomyopathy. Lab. Investig. 2018, 98, 1291–1299.
- Lowe, G.D. Fibrinogen Assays for Cardiovascular Risk Assessment. Clin. Chem. 2010, 56, 693–695.
- Danesh, J.; Lewington, S.; Thompson, S.G.; Lowe, G.D.O.; Collins, R.; Kostis, J.B.; Wilson, A.C.; Folsom, A.R.; Wu, K.; Benderly, M.; et al. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: An individual participant meta-analysis. JAMA 2005, 294, 1799–1809.
- The Emerging Risk Factors Collaboration. C-Reactive Protein, Fibrinogen, and Cardiovascular Disease Prediction. N. Engl. J. Med. 2012, 367, 1310–1320.
- Appiah, D.; Schreiner, P.J.; MacLehose, R.F.; Folsom, A.R. Association of Plasma γ′ Fibrinogen with Incident Cardiovascular Disease: The Atherosclerosis Risk in Communities (ARIC) Study. ATVB 2015, 35, 2700–2706.
- Becatti, M.; Marcucci, R.; Bruschi, G.; Taddei, N.; Bani, D.; Gori, A.M.; Giusti, B.; Gensini, G.F.; Abbate, R.; Fiorillo, C. Oxidative Modification of Fibrinogen Is Associated with Altered Function and Structure in the Subacute Phase of Myocardial Infarction. ATVB 2014, 34, 1355–1361.
- Klip, I.T.; Comin-Colet, J.; Voors, A.A.; Ponikowski, P.; Enjuanes, C.; Banasiak, W.; Lok, D.J.; Rosentryt, P.; Torrens, A.; Polonski, L.; et al. Iron deficiency in chronic heart failure: An international pooled analysis. Am. Heart J. 2013, 165, 575–582.e3.
- Comín-Colet, J.; Ruiz, S.; Cladellas, M.; Rizzo, M.; Torres, A.; Bruguera, J. A Pilot Evaluation of the Long-term Effect of Combined Therapy with Intravenous Iron Sucrose and Erythropoietin in Elderly Patients with Advanced Chronic Heart Failure and Cardio-Renal Anemia Syndrome: Influence on Neurohormonal Activation and Clinical Outcomes. J. Card. Fail. 2009, 15, 727–735.
- Jankowska, E.A.; Rozentryt, P.; Witkowska, A.; Nowak, J.; Hartmann, O.; Ponikowska, B.; Borodulin-Nadzieja, L.; Von Haehling, S.; Doehner, W.; Banasiak, W.; et al. Iron Deficiency Predicts Impaired Exercise Capacity in Patients with Systolic Chronic Heart Failure. J. Card. Fail. 2011, 17, 899–906.
- Enjuanes, C.; Klip, I.T.; Bruguera, J.; Cladellas, M.; Ponikowski, P.; Banasiak, W.; Van Veldhuisen, D.J.; Van Der Meer, P.; Jankowska, E.A.; Comín-Colet, J. Iron deficiency and health-related quality of life in chronic heart failure: Results from a multicenter European study. Int. J. Cardiol. 2014, 174, 268–275.
- Van Veldhuisen, D.J.; Anker, S.D.; Ponikowski, P.; Macdougall, I.C. Anemia and iron deficiency in heart failure: Mechanisms and therapeutic approaches. Nat Rev Cardiol 2011, 8, 485–493.
- Gettins, P.G.W. Serpin Structure, Mechanism, and Function. Chem. Rev. 2002, 102, 4751–4804.
- Izquierdo, I.; Rosa, I.; Bravo, S.B.; Guitián, E.; Pérez-Serra, A.; Campuzano, O.; Brugada, R.; Mangas, A.; García, Á.; Toro, R. Proteomic identification of putative biomarkers for early detection of sudden cardiac death in a family with a LMNA gene mutation causing dilated cardiomyopathy. J. Proteom. 2016, 148, 75–84.
- Wu, T.; Chen, Y.; Du, Y.; Tao, J.; Zhou, Z.; Yang, Z. Serum Exosomal MiR-92b-5p as a Potential Biomarker for Acute Heart Failure Caused by Dilated Cardiomyopathy. Cell Physiol. Biochem. 2018, 46, 1939–1950.
- Fu, X.; Mishra, R.; Chen, L.; Arfat, M.Y.; Sharma, S.; Kingsbury, T.; Gunasekaran, M.; Saha, P.; Hong, C.; Yang, P.; et al. Exosomes mediated fibrogenesis in dilated cardiomyopathy through a MicroRNA pathway. iScience 2023, 26, 105963.
- Vandergriff, A.; Huang, K.; Shen, D.; Hu, S.; Hensley, M.T.; Caranasos, T.G.; Qian, L.; Cheng, K. Targeting regenerative exosomes to myocardial infarction using cardiac homing peptide. Theranostics 2018, 8, 1869–1878.
- Sun, X.; Shan, A.; Wei, Z.; Xu, B. Intravenous mesenchymal stem cell-derived exosomes ameliorate myocardial inflammation in the dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2018, 503, 2611–2618.
- Ni, J.; Liu, Y.; Wang, K.; Wu, M.; Kang, L.; Sha, D.; Xu, B.; Gu, R. Trophoblast Stem-Cell-Derived Exosomes Improve Doxorubicin-Induced Dilated Cardiomyopathy by Modulating the let-7i/YAP Pathway. Mol. Ther.-Nucleic Acids 2020, 22, 948–956.
- Zhang, W.; Chen, Z.; Qiao, S.; Chen, S.; Zheng, H.; Wei, X.; Li, Q.; Xu, B.; Huang, W. The effects of extracellular vesicles derived from Krüppel-Like Factor 2 overexpressing endothelial cells on the regulation of cardiac inflammation in the dilated cardiomyopathy. J. Nanobiotechnol. 2022, 20, 76.
- World Health Organization G. The top 10 Causes of Death . WHO. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 8 December 2023).
- Kannel, W.B. Diabetes and Cardiovascular Disease: The Framingham Study. JAMA 1979, 241, 2035.
- Jia, G.; Whaley-Connell, A.; Sowers, J.R. Diabetic cardiomyopathy: A hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia 2018, 61, 21–28.
- Seferović, P.M.; Paulus, W.J. Clinical diabetic cardiomyopathy: A two-faced disease with restrictive and dilated phenotypes. Eur. Heart J. 2015, 36, 1718–1727.
- Wang, L.; Cai, Y.; Jian, L.; Cheung, C.W.; Zhang, L.; Xia, Z. Impact of peroxisome proliferator-activated receptor-α on diabetic cardiomyopathy. Cardiovasc. Diabetol. 2021, 20, 2.
- De Gonzalo-Calvo, D.; Van Der Meer, R.W.; Rijzewijk, L.J.; Smit, J.W.A.; Revuelta-Lopez, E.; Nasarre, L.; Escola-Gil, J.C.; Lamb, H.J.; Llorente-Cortes, V. Serum microRNA-1 and microRNA-133a levels reflect myocardial steatosis in uncomplicated type 2 diabetes. Sci. Rep. 2017, 7, 47.
- Huang, J.-P.; Chang, C.-C.; Kuo, C.-Y.; Huang, K.-J.; Sokal, E.M.; Chen, K.-H.; Hung, L.-M. Exosomal microRNAs miR-30d-5p and miR-126a-5p Are Associated with Heart Failure with Preserved Ejection Fraction in STZ-Induced Type 1 Diabetic Rats. Int. J. Mol. Sci. 2022, 23, 7514.
- Watson, C.J.; Gupta, S.K.; O’Connell, E.; Thum, S.; Glezeva, N.; Fendrich, J.; Gallagher, J.; Ledwidge, M.; Grote-Levi, L.; McDonald, K.; et al. MicroRNA signatures differentiate preserved from reduced ejection fraction heart failure. Eur. J. Heart Fail. 2015, 17, 405–415.
- Vegter, E.L.; Van Der Meer, P.; De Windt, L.J.; Pinto, Y.M.; Voors, A.A. MicroRNAs in heart failure: From biomarker to target for therapy. Eur. J Heart Fail. 2016, 18, 457–468.
- Li, J.; Salvador, A.M.; Li, G.; Valkov, N.; Ziegler, O.; Yeri, A.; Yang Xiao, C.; Meechoovet, B.; Alsop, E.; Rodosthenous, R.S.; et al. Mir-30d Regulates Cardiac Remodeling by Intracellular and Paracrine Signaling. Circ. Res. 2021, 128, E1–E23.
- Potus, F.; Ruffenach, G.; Dahou, A.; Thebault, C.; Breuils-Bonnet, S.; Tremblay, È.; Nadeau, V.; Paradis, R.; Graydon, C.; Wong, R.; et al. Downregulation of MicroRNA-126 Contributes to the Failing Right Ventricle in Pulmonary Arterial Hypertension. Circulation 2015, 132, 932–943.
- Hu, J.; Wang, S.; Xiong, Z.; Cheng, Z.; Yang, Z.; Lin, J.; Wang, T.; Feng, X.; Gao, E.; Wang, H.; et al. Exosomal Mst1 transfer from cardiac microvascular endothelial cells to cardiomyocytes deteriorates diabetic cardiomyopathy. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 3639–3649.
- Zhang, Y.; Zhu, Z.; Wang, T.; Dong, Y.; Fan, Y.; Sun, D. TGF-β1-containing exosomes from cardiac microvascular endothelial cells mediate cardiac fibroblast activation under high glucose conditions. Biochem. Cell Biol. 2021, 99, 693–699.
- Richter, K.; Haslbeck, M.; Buchner, J. The Heat Shock Response: Life on the Verge of Death. Mol. Cell 2010, 40, 253–266.
- Wang, X.; Gu, H.; Huang, W.; Peng, J.; Li, Y.; Yang, L.; Qin, D.; Essandoh, K.; Wang, Y.; Peng, T.; et al. Hsp20-Mediated Activation of Exosome Biogenesis in Cardiomyocytes Improves Cardiac Function and Angiogenesis in Diabetic Mice. Diabetes 2016, 65, 3111–3128.
- Lin, Y.; Zhang, F.; Lian, X.-F.; Peng, W.-Q.; Yin, C.-Y. Mesenchymal stem cell-derived exosomes improve diabetes mellitus-induced myocardial injury and fibrosis via inhibition of TGF-β1/Smad2 signaling pathway. Cell Mol. Biol. (Noisy-le-grand) 2019, 65, 123–126.
- Singla, R.; Garner, K.H.; Samsam, M.; Cheng, Z.; Singla, D.K. Exosomes derived from cardiac parasympathetic ganglionic neurons inhibit apoptosis in hyperglycemic cardiomyoblasts. Mol. Cell Biochem. 2019, 462, 1–10.
- Zhen, J.; Bai, J.; Liu, J.; Men, H.; Yu, H. Ginsenoside RG1-induced mesenchymal stem cells alleviate diabetic cardiomyopathy through secreting exosomal circNOTCH1 to promote macrophage M2 polarization. Phytother. Res. 2023.
More