Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Everton Freitas de Morais and Version 2 by Sirius Huang.
Oral squamous cell carcinoma (OSCC) is the most common and lethal type of head and neck cancer in the world. Variable response and acquisition of resistance to traditional therapies show that it is essential to develop novel strategies that can provide better outcomes for the patient. Activation of cell death pathways, such as the emerging forms of non-apoptotic programmed cell death, including ferroptosis, pyroptosis, necroptosis, NETosis, parthanatos, mitoptosis and paraptosis, may represent clinically relevant novel therapeutic opportunities.
- emerging types of cell death
- tumor microenvironment
- oral cancer
- ferroptosis
- pyroptosis
- necroptosis
1. Introduction
Among the hallmarks of cancer, the acquisition of resistance to cell death plays an important role in cancer initiation and progression to high-grade malignant states, which are frequently unresponsive to conventional anti-cancer therapies [1]. In recent years, there has been a significant improvement in the understanding of different mechanisms of cell death beyond the traditionally observed apoptosis and necrosis [2][3][2,3]. Alternative cell death pathways, collectively called programmed cell death (PCD), have been identified in a variety of pathological processes, including oral cancer [4]. These pathways not only contribute to the understanding of cancer pathophysiology [5] but also reveal an intricate balance between molecules involved in cell survival and death, with significant implications, for example, as prognostic biomarkers and in the development of new therapeutic strategies that are beginning to be explored [6].
While malignant cells develop strategies to escape or limit conventional cell death pathways, understanding of their ability to escape death by other mechanisms is much more limited [5]. Since 2018, the Nomenclature Committee on Cellular Death has classified PCD into 12 subtypes of death that differ in molecular mechanisms but may share small similarities in their morphological characteristics, ranging from a necrotic profile, that is, unprogrammed and with a disordered appearance, to an apoptotic profile with an organized profile [2][7][2,7]. However, an update proposed by Yan, Elbadawi and Efferth [3] divided cell deaths into three large groups based on activation of specific signaling pathways: (1) Non-programmed cell death (NPCD) or necrosis, (2) apoptotic programmed cell death (APCD) and (3) non-apoptotic programmed cell death (NAPCD), which differs from the apoptotic form because it does not maintain the integrity of the cell membrane and is independent of caspases. Similar to apoptosis, NAPCD has a highly regulated molecular machinery that can be targeted or modulated by molecular strategies [2].
The most studied emerging NAPCD types in oncology are ferroptosis, pyroptosis and necroptosis [8]. Ferroptosis involves the accumulation of lipid peroxides due to disrupted cellular antioxidant defenses, leading to oxidative stress-induced cell death [9]. Cancer cells can escape ferroptosis by enhancing antioxidant defenses and modifying lipid metabolism, enabling them to survive and proliferate despite conditions that typically trigger ferroptotic cell death [10]. Meanwhile, during pyroptosis and necroptosis, the intracellular content is expelled from the cell through membrane pores formed by proteins such as those from the Gasdermin family (GSDM) in pyroptosis and mixed lineage kinase domain-like (MLKL) in necroptosis, resulting in recruitment of inflammatory cells [11][12][11,12]. The pro-inflammatory environment induced by the extravasation of intracellular contents may promote the transformation and progression of tumor cells [13][14][13,14]. However, the role of NAPCD in cancer progression is only partially understood, and the literature is conflicting.
2. Ferroptosis
Ferroptosis is a distinguished type of NAPCD characterized by iron-dependent lipid peroxide accumulation, particularly of polyunsaturated fatty acids [15][16][97,98]. The research landscape of this type of cell death and its implications in diseases is relatively recent [17][18][99,100] but promising for increased knowledge on mechanisms and therapeutic strategies in cancer [17][19][99,101]. Ferroptosis can occur through two major pathways: the extrinsic pathway or transporter-dependent, and the intrinsic or enzyme-regulated pathway [17][20][99,102] (Figure 1). Despite being distinct pathways, it is important to highlight that one can influence the other, as both rely on iron metabolism and glutathione (GSH)-dependent antioxidant mechanisms [21][103]. The extrinsic mechanism depends on the balance of iron and amino acid transport across the cell membrane [20][102]. A higher intracellular iron level increases the production of reactive oxygen species (ROS) through the Fenton reaction [22][104]. Moreover, inhibition of the Xc-system reduces cystine uptake, which is essential for synthesizing GSH [20][102]. The depletion of GSH reduces the cell’s antioxidant defenses and favors lipid peroxidation. In turn, the intrinsic pathway is mainly induced by inhibiting glutathione peroxidase 4 (GPX4), an enzyme that plays a pivotal role in reducing lipid hydroperoxides to non-toxic lipid alcohols using GSH as a cofactor [23][105]. The inhibition of GPX4 activity occurs in an unchecked accumulation of lipid hydroperoxides, leading to cellular damage and eventual ferroptosis cell death [20][23][102,105].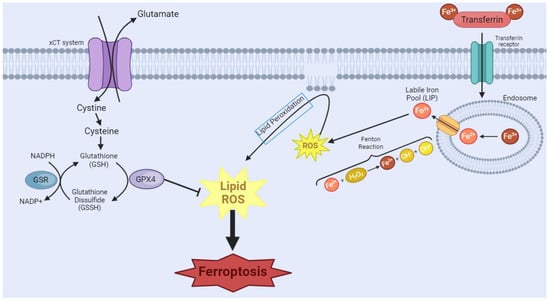
Figure 1. Ferroptosis manifests through two primary pathways. In the first pathway, transferrin (TRF1), a crucial player in iron homeostasis, binds to Fe3+, forming a complex, which then binds to the transferrin receptor. Endocytosis of this complex occurs, leading to the formation of the endosome. Within the endosome, there will be acidification of the environment, causing the dissociation of the complex and the reduction of Fe3+ to Fe2+. This Fe2+ is released through the endosomal membrane. However, surplus free iron ions form a labile iron pool (LIP), which partakes in Fenton’s reactions, causing the generation of the reactive oxygen species (ROS) free radical hydroxyl. This cascade of events culminates in the initiation of lipoperoxidation, a critical step in the ferroptotic process. In the second pathway, System Xc-, a cystine/glutamate antiporter, imports extracellular cystine. Intracellularly, cystine undergoes conversion into glutathione (GSH), a vital antioxidant. The availability of GSH is integral to the function of glutathione peroxidase 4 (GPX4), which tackles intracellular lipid peroxides, preventing ferroptosis. However, the inhibition of GPX4 leads to a disruption in this protective mechanism, resulting in an augmented presence of ROS and ultimately contributing to the progression of ferroptosis. [Image was created using Biorender.com (accessed on 16 November 2023)].
In summary, several studies have shown promising results in using the existent ferroptosis-related cellular machinery as potential therapeutic strategies. Both knockdown strategies targeting key proteins involved in promoting/regulating this type of cell death and application of compounds with activity to induce ferroptosis of the OSCC cells were used. Additionally, the characterization of several gene signatures of ferroptosis-associated genes for both prognostication and response to treatment is relevant, but further verification in large-scale clinical studies is required. Thereafter, the ongoing exploration of ferroptosis in oral cancer not only deepens our understanding of cancer mechanisms but also holds the potential to translate this knowledge to improve patient outcomes.
 Inflammation is a critical component of tumor progression [88][146], and inflammation intensified by chemotherapy can lead to therapy failure and metastasis [89][147]. In the Nod-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome is one of the critical components of the innate immune system and plays an important role in cancer [90][91][148,149]. Many factors can activate NLRP3 inflammasomes, including potassium efflux, intracellular calcium, endoplasmic reticulum (ER) stress and ROS [91][149]. Chronic inflammation has the potential to impact every phase of the carcinogenic process, increasing the risk of tumorigenesis with prolonged exposure to an inflammatory milieu [92][150]. Pyroptosis, as a form of lytic cell death, amplifies the release of mature interleukin-1 (IL-1) and interleukin-18 (IL-18), potentially influencing the development of cancer [93][151]. Furthermore, pyroptosis serves as the mechanism for inflammatory cell death in cancer cells, thereby restraining the proliferation and migration of these cancer cells [78][79][94][95][96][97][98][28,33,68,69,70,78,79]. Consequently, pyroptosis assumes a dual role, both promoting and inhibiting tumorigenesis [99][152]. Previous findings revealed that 5-fluorouracil (5-FU) treatment increased NLRP3 expression in OSCC, which mediated drug resistance. It was also proven that NLRP3 could promote tumor growth and metastasis in OSCC [97][100][101][64,69,153]. Activation of pyroptosis has also been directly associated with increased chemoresistance to cisplatin and 5-FU treatment [100][64], and inhibition of pyroptosis has been associated with increased sensitivity of neoplastic cells to cisplatin treatment [102][66].
Methods of pyroptosis inhibition are gaining the attention of the scientific community [97][103][104][105][65,69,74,154]. Yang et al. [97][69] highlighted that extracellular vesicles derived from bitter melon led to a significant decrease in the expression of NLRP3, reducing OSCC resistance to 5-FU treatment. The study developed by Yue et al. [103][65] evaluated the effect of anthocyanin on OSCC. Anthocyanin reduced the viability of OSCC cells and inhibited migration and invasion capacity, concomitantly increasing pyroptosis. Simultaneously, activation of pyroptosis was associated with increased expression of NLRP3, caspase-1 and interleukin-1β (IL-1β). After administration of caspase-1 inhibitors, anthocyanin-activated pyroptosis was suppressed, and cell viability, migration and invasion rates increased concomitantly. In vitro studies in monoculture do not show the real dimension of the role of pyroptosis in OSCC. Therefore, conflicting results can be seen depending on the methodology used in different studies. The poor prognosis of pyroptosis is associated with its ability to activate inflammation; however, the development of in vitro and in vivo studies capable of more broadly evaluating the tumor microenvironment and all mechanisms triggered by the activation of pyroptosis should be encouraged. The inflammation associated with pyroptosis can lead to the recruitment of immune cells and other factors that support tumor growth and metastasis [105][154]. The pro-inflammatory environment can also contribute to resistance to therapy and promote angiogenesis, which is the formation of new blood vessels that supply nutrients to the tumor. The study developed by Xin et al. [104][74] used The Cancer Genome Atlas (TCGA) dataset to investigate the predictive value of pyroptosis-related lncRNAs in the prognosis of OSCC. The authors identified eight pyroptosis-related lncRNAs associated with overall survival in patients with OSCC by multivariate regression analysis.
Taken together, the analysis indicates that the number of studies exploring pyroptosis in OSCC is still restricted, limiting our knowledge of the mechanisms of how molecules related to this type of cell death affect OSCC cells. However, the studies highlighted the potential of pyroptosis in the control of OSCC development and progression and described drugs and molecules related to both blockage and induction of it. Moreover, two studies demonstrated that pyroptosis gene clusters are correlated with clinical characteristics, infiltration of immune cells, susceptibility to chemotherapy and immunotherapy, and prognosis of patients with OSCC [104][106][74,75]. It will be important to validate these risk models and to explore potential drugs that could induce cell death through pyroptosis and concomitantly inhibit the pro-tumor inflammatory response in OSCCs.
Inflammation is a critical component of tumor progression [88][146], and inflammation intensified by chemotherapy can lead to therapy failure and metastasis [89][147]. In the Nod-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome is one of the critical components of the innate immune system and plays an important role in cancer [90][91][148,149]. Many factors can activate NLRP3 inflammasomes, including potassium efflux, intracellular calcium, endoplasmic reticulum (ER) stress and ROS [91][149]. Chronic inflammation has the potential to impact every phase of the carcinogenic process, increasing the risk of tumorigenesis with prolonged exposure to an inflammatory milieu [92][150]. Pyroptosis, as a form of lytic cell death, amplifies the release of mature interleukin-1 (IL-1) and interleukin-18 (IL-18), potentially influencing the development of cancer [93][151]. Furthermore, pyroptosis serves as the mechanism for inflammatory cell death in cancer cells, thereby restraining the proliferation and migration of these cancer cells [78][79][94][95][96][97][98][28,33,68,69,70,78,79]. Consequently, pyroptosis assumes a dual role, both promoting and inhibiting tumorigenesis [99][152]. Previous findings revealed that 5-fluorouracil (5-FU) treatment increased NLRP3 expression in OSCC, which mediated drug resistance. It was also proven that NLRP3 could promote tumor growth and metastasis in OSCC [97][100][101][64,69,153]. Activation of pyroptosis has also been directly associated with increased chemoresistance to cisplatin and 5-FU treatment [100][64], and inhibition of pyroptosis has been associated with increased sensitivity of neoplastic cells to cisplatin treatment [102][66].
Methods of pyroptosis inhibition are gaining the attention of the scientific community [97][103][104][105][65,69,74,154]. Yang et al. [97][69] highlighted that extracellular vesicles derived from bitter melon led to a significant decrease in the expression of NLRP3, reducing OSCC resistance to 5-FU treatment. The study developed by Yue et al. [103][65] evaluated the effect of anthocyanin on OSCC. Anthocyanin reduced the viability of OSCC cells and inhibited migration and invasion capacity, concomitantly increasing pyroptosis. Simultaneously, activation of pyroptosis was associated with increased expression of NLRP3, caspase-1 and interleukin-1β (IL-1β). After administration of caspase-1 inhibitors, anthocyanin-activated pyroptosis was suppressed, and cell viability, migration and invasion rates increased concomitantly. In vitro studies in monoculture do not show the real dimension of the role of pyroptosis in OSCC. Therefore, conflicting results can be seen depending on the methodology used in different studies. The poor prognosis of pyroptosis is associated with its ability to activate inflammation; however, the development of in vitro and in vivo studies capable of more broadly evaluating the tumor microenvironment and all mechanisms triggered by the activation of pyroptosis should be encouraged. The inflammation associated with pyroptosis can lead to the recruitment of immune cells and other factors that support tumor growth and metastasis [105][154]. The pro-inflammatory environment can also contribute to resistance to therapy and promote angiogenesis, which is the formation of new blood vessels that supply nutrients to the tumor. The study developed by Xin et al. [104][74] used The Cancer Genome Atlas (TCGA) dataset to investigate the predictive value of pyroptosis-related lncRNAs in the prognosis of OSCC. The authors identified eight pyroptosis-related lncRNAs associated with overall survival in patients with OSCC by multivariate regression analysis.
Taken together, the analysis indicates that the number of studies exploring pyroptosis in OSCC is still restricted, limiting our knowledge of the mechanisms of how molecules related to this type of cell death affect OSCC cells. However, the studies highlighted the potential of pyroptosis in the control of OSCC development and progression and described drugs and molecules related to both blockage and induction of it. Moreover, two studies demonstrated that pyroptosis gene clusters are correlated with clinical characteristics, infiltration of immune cells, susceptibility to chemotherapy and immunotherapy, and prognosis of patients with OSCC [104][106][74,75]. It will be important to validate these risk models and to explore potential drugs that could induce cell death through pyroptosis and concomitantly inhibit the pro-tumor inflammatory response in OSCCs.
 The formation of an MLKL oligomer opens a pore in the membrane, allowing the entrance of ROS and DAMPs [112][160]. In this manner, death by necroptosis, despite being activated by specific signals, leads to a similar end as necrosis and ferroptosis, triggering the entrance of ROS in the cell and leading to membrane damage [112][113][160,161]. To allow for necroptosis to occur, the apoptotic pathway must be impaired or damaged [107][114][115][155,162,163], which can be quite common in cancer since the inhibition of healthy apoptosis as a control allows for the unchecked reproduction of damaged cells [116][164]. This phenomenon holds significant implications for oral cancer, where dysregulation of cell death mechanisms can tip the balance in favor of tumor progression.
In this context, necroptosis, which is characterized by a regulated inflammatory response, becomes a last-resort mechanism to eliminate aberrant cells [117][118][165,166]. However, cancer cells can hijack this mechanism to promote their survival and evade the body’s natural defenses, using pro-tumoral inflammation to their advantage [117][165]. Additionally, necroptosis can generate an immunosuppressive tumor microenvironment, which may further contribute to cancer cell survival and progression [119][167]. It comes as no surprise that the expression level of key mediators of necroptosis is elevated in cancer [117][120][165,168], indicating that necroptosis may play a role in promoting oncogenesis and cancer metastasis [121][169]. However, it is possible to interfere in this process by targeting necroptosis as an ally in halting cancer progression and survival, as has been evaluated in oral cancer studies, by targeting focal adhesion molecules [122][123][170,171] or even caspase-8 itself, which is responsible for deciding the pathway outcome of apoptosis versus necrosis [122][124][170,172]. In the context of OSCC, studies have explored the induction of necroptosis using different agents, such as Obatoclax [125][84]. This agent targets members of the BCL-2 family, specifically the myeloid cell leukemia sequence 1 (MCL-1). The study suggests that Obatoclax induces cell death in OSCC cells through autophagy-dependent necroptosis [126][173], with mitochondrial stress and dysfunction as detectable upstream events. Additionally, capsaicin was found to inhibit cell proliferation and induce endoplasmic reticulum stress and autophagy in oral cancer [127][87]. This mechanism negatively regulates ribophorin II, impairing P-glycoprotein functions and sensitizing cells to anticancer therapy [128][174]. The association of capsaicin with anticancer agents promotes necroptosis rather than apoptosis, showcasing a unique pathway for inhibiting OSCC cell viability. Similarly, chelerythrine chloride (CS) demonstrated necroptosis induction in OSCC, impairing cell proliferation and inducing morphological alterations in a dose-dependent manner, such as membrane rupture, and dose-dependent cell death [127][87]. Moreover, the development of targeted delivery systems such as PLGA-Dtx (poly-lactic-co-glycolic acid nanoparticles containing docetaxel) has shown enhanced efficacy in inhibiting cancer cell proliferation. This strategy induced both apoptosis and necroptosis in oral cancer cells [129][89]. These results altogether suggest a potential role for these compounds in triggering necroptosis in OSCC cells.
Studies on necroptosis-related genes may reveal potential targets, either enhancers or inhibitors of this NAPCD. HNSCC studies have associated CASP8 mutations with radioresistance and poor survival outcomes [130][86], as is the case for OSCC [131][175]. In this manner, knockdown of CASP8 enhances the radiosensitizing effects of certain compounds through the induction of necroptosis. Additionally, as seen in other cell death mechanisms, the investigation of necroptosis-related genes that may be relevant in prognosis prediction has also been explored. Bioinformatic analyses and in vitro experiments identified six genes (hypoxanthine phosphoribosyltransferase 1-HPRT1, PGAM family member 5, mitochondrial serine/threonine protein phosphatase-PGAM5, BH3 interacting domain death agonist-BID, survival of motor neuron 1, telomeric-SMN1, FADD, and KIAA1191) contributing to OSCC development, metastasis, and immune modulation. These genes may play a role in the regulation of cell death pathways, including necroptosis [132][90]. Moreover, a study in HNSCC emphasized the prevalence of necroptosis and its association with poor overall survival and progression-free survival. Approximately half of the necrosis in HNSCC was attributed to necroptosis, indicating its significance as an independent risk factor for adverse clinical outcomes [133][85].
In summary, necroptosis induction is a promising alternative in the treatment of oral cancers, and the expression of necroptosis-related genes depicts prognostic potential for predicting OSCC outcomes. However, one of the main challenges in inducing necroptosis as a cancer treatment strategy remains in establishing specificity in a manner by which little toxicity is archived [124][172].
The formation of an MLKL oligomer opens a pore in the membrane, allowing the entrance of ROS and DAMPs [112][160]. In this manner, death by necroptosis, despite being activated by specific signals, leads to a similar end as necrosis and ferroptosis, triggering the entrance of ROS in the cell and leading to membrane damage [112][113][160,161]. To allow for necroptosis to occur, the apoptotic pathway must be impaired or damaged [107][114][115][155,162,163], which can be quite common in cancer since the inhibition of healthy apoptosis as a control allows for the unchecked reproduction of damaged cells [116][164]. This phenomenon holds significant implications for oral cancer, where dysregulation of cell death mechanisms can tip the balance in favor of tumor progression.
In this context, necroptosis, which is characterized by a regulated inflammatory response, becomes a last-resort mechanism to eliminate aberrant cells [117][118][165,166]. However, cancer cells can hijack this mechanism to promote their survival and evade the body’s natural defenses, using pro-tumoral inflammation to their advantage [117][165]. Additionally, necroptosis can generate an immunosuppressive tumor microenvironment, which may further contribute to cancer cell survival and progression [119][167]. It comes as no surprise that the expression level of key mediators of necroptosis is elevated in cancer [117][120][165,168], indicating that necroptosis may play a role in promoting oncogenesis and cancer metastasis [121][169]. However, it is possible to interfere in this process by targeting necroptosis as an ally in halting cancer progression and survival, as has been evaluated in oral cancer studies, by targeting focal adhesion molecules [122][123][170,171] or even caspase-8 itself, which is responsible for deciding the pathway outcome of apoptosis versus necrosis [122][124][170,172]. In the context of OSCC, studies have explored the induction of necroptosis using different agents, such as Obatoclax [125][84]. This agent targets members of the BCL-2 family, specifically the myeloid cell leukemia sequence 1 (MCL-1). The study suggests that Obatoclax induces cell death in OSCC cells through autophagy-dependent necroptosis [126][173], with mitochondrial stress and dysfunction as detectable upstream events. Additionally, capsaicin was found to inhibit cell proliferation and induce endoplasmic reticulum stress and autophagy in oral cancer [127][87]. This mechanism negatively regulates ribophorin II, impairing P-glycoprotein functions and sensitizing cells to anticancer therapy [128][174]. The association of capsaicin with anticancer agents promotes necroptosis rather than apoptosis, showcasing a unique pathway for inhibiting OSCC cell viability. Similarly, chelerythrine chloride (CS) demonstrated necroptosis induction in OSCC, impairing cell proliferation and inducing morphological alterations in a dose-dependent manner, such as membrane rupture, and dose-dependent cell death [127][87]. Moreover, the development of targeted delivery systems such as PLGA-Dtx (poly-lactic-co-glycolic acid nanoparticles containing docetaxel) has shown enhanced efficacy in inhibiting cancer cell proliferation. This strategy induced both apoptosis and necroptosis in oral cancer cells [129][89]. These results altogether suggest a potential role for these compounds in triggering necroptosis in OSCC cells.
Studies on necroptosis-related genes may reveal potential targets, either enhancers or inhibitors of this NAPCD. HNSCC studies have associated CASP8 mutations with radioresistance and poor survival outcomes [130][86], as is the case for OSCC [131][175]. In this manner, knockdown of CASP8 enhances the radiosensitizing effects of certain compounds through the induction of necroptosis. Additionally, as seen in other cell death mechanisms, the investigation of necroptosis-related genes that may be relevant in prognosis prediction has also been explored. Bioinformatic analyses and in vitro experiments identified six genes (hypoxanthine phosphoribosyltransferase 1-HPRT1, PGAM family member 5, mitochondrial serine/threonine protein phosphatase-PGAM5, BH3 interacting domain death agonist-BID, survival of motor neuron 1, telomeric-SMN1, FADD, and KIAA1191) contributing to OSCC development, metastasis, and immune modulation. These genes may play a role in the regulation of cell death pathways, including necroptosis [132][90]. Moreover, a study in HNSCC emphasized the prevalence of necroptosis and its association with poor overall survival and progression-free survival. Approximately half of the necrosis in HNSCC was attributed to necroptosis, indicating its significance as an independent risk factor for adverse clinical outcomes [133][85].
In summary, necroptosis induction is a promising alternative in the treatment of oral cancers, and the expression of necroptosis-related genes depicts prognostic potential for predicting OSCC outcomes. However, one of the main challenges in inducing necroptosis as a cancer treatment strategy remains in establishing specificity in a manner by which little toxicity is archived [124][172].
3. Pyroptosis
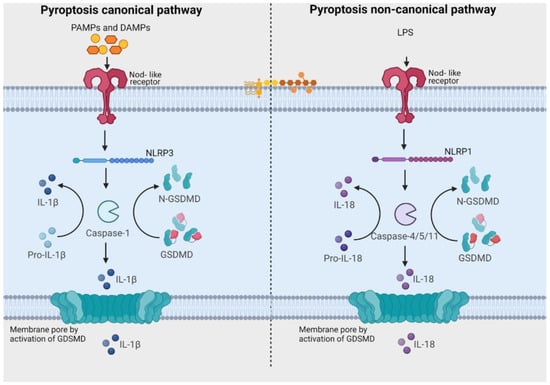
Pyroptosis, an inflammatory type of caspase-mediated cell death, can modulate the immunogenic potential of specific cancers [75][82]. The role of pyroptosis in OSCC is an area of ongoing research, and the mechanisms and implications are not fully understood, but there is evidence to suggest that pyroptosis may play a role in both development and progression of OSCC [75][76][77][78][79][80][81][82][76,77,78,79,80,81,82,83]. Pyroptosis derives its name from the combination of “pyro” and “ptosis”. “Pyro” signifies fire, highlighting its inflammatory properties, while “ptosis” refers to falling, which aligns with other forms of programmed cell death [83][141]. There are notable similarities between pyroptosis and apoptosis, including features like DNA damage and chromatin condensation [84][142]. Interestingly, pyroptotic cells exhibit swelling and numerous bubble-like protrusions on the cellular membrane before rupture, a phenomenon reminiscent of membrane blebbing observed in apoptosis. Pyroptosis was officially defined as Gasdermin-mediated programmed cell death in 2015 [83][141]. The Gasdermin superfamily in humans includes Gasdermin A/B/C/D (GSDMA/B/C/D), Gasdermin E (GSDME, also known as DFNA5), and DFNB59 (Pejvakin, PJVK). Inflammasomes are responsible for initiating pyroptosis via two distinct pathways (Figure 2). The canonical inflammasome pathway is reliant on the activation of caspase-1, and the noncanonical inflammasome pathway involves the activation of caspase-4, caspase-5, or caspase-11 [85][143]. Furthermore, certain studies have demonstrated that proapoptotic caspase-3 activation can also initiate pyroptosis by cleaving GSDME [86][87][144,145].Figure 2. Pathways related to activation of pyroptosis. In the canonical signaling pathway, intracellular sensors Nod-like receptor family, pyrin domain containing 1 (NLRP1), 3 (NLRP3), 4 (NLRC4), absent in melanoma 2 (AIM2) and other inflammasome sensors are responsible for detecting microbial signals. Upon detection, they initiate a response by recruiting the adaptor protein ASC (apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain), which subsequently recruits pro-caspase-1. Once activated, caspase-1 cleaves Gasdermin D (GSDMD), generating GSDMD-NT fragments. These GSDMD-NT fragments create pores in the plasma membrane that are associated with phosphoinositides. Simultaneously, caspase-1 itself undergoes cleavage, giving rise to caspase-1 P10/P20 and P33/P10 tetramers. These tetramers play a crucial role in the maturation of pro-interleukin-18 (IL-18) and pro-interleukin-1β (IL-1β) into their active forms, IL-18 and IL-1β. These mature cytokines are subsequently released into the extracellular matrix, leading to the initiation of inflammatory responses. In the noncanonical pathway, the presence of lipopolysaccharides (LPS) from Gram-negative bacteria triggers the activation of caspase-4 and caspase-5 (in humans) or caspase-11 (in mice). These caspases, in turn, cleave GSDMD, forming pores in the plasma membrane. These GSDMD pores permit the release of potassium ions, which further activate the NLRP3 inflammasome and contribute to the maturation of IL-1β and IL-18. Additionally, GSDMD pores release mature cytokines, ultimately leading to pyroptosis. [Image was created using Biorender.com (accessed on 13 December 2023)].
4. Necroptosis
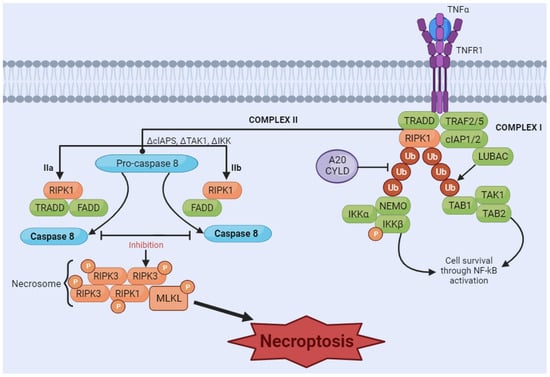
Necroptosis is considered a programmed form of necrosis mediated by receptor-interacting protein kinase 1 (RIPK1) and receptor-interacting protein kinase 3 (RIPK3) [107][108][155,156]. The stimulus is initiated by tumor necrosis factor (TNF) binding to its receptors [109][110][157,158]. The interaction of ligand and receptor leads to the formation of a signaling complex, which may include adaptor proteins such as Fas-associated protein with death domain (FADD) and TNF receptor-associated death domain (TRADD). RIPK1 is recruited to the signaling complex and is activated by phosphorylation. Depending on cellular conditions, RIPK1 can associate with caspases, promoting apoptosis, or with RIPK3, initiating the necroptosis pathway. If the necroptosis pathway is activated, RIPK1 recruits RIPK3 and the protein MLKL, forming the necrosome complex. RIPK3 phosphorylates MLKL, activating it. Activated MLKL oligomerizes and translocates to the plasma membrane, where it causes damage, leading to membrane rupture and necroptosis (Figure 3) [111][159].Figure 3. Necroptosis signaling pathway. The binding of tumor necrosis factor (TNF) induces the formation of the membrane-associated complex I, composed of TNF receptor-associated death domain (TRADD), TNF receptor-associated factor 2 (TRAF2), cellular inhibitor of apoptosis protein 1 and 2 (cIAP1/2), receptor-interacting serine/threonine kinase 1 (RIPK1), and LUBAC, an E3 ubiquitin ligase. cIAP1/2 and LUBAC induce the polyubiquitination of RIPK1, recruiting the IκB kinase (IkappaB kinase) complex (IKKa, IKKB, and NEMO) and TGF-β-activated kinase 1 (TAK1) complex (TAK1, TAB1, and TAB2). These two complexes can eventually lead to the activation of the NF-κB pathway and cell survival. A20CYLD promotes the deubiquitination of RIPK1, inducing the dissociation of TRADD and RIPK1 from TNFR1 and leading to the formation of complex IIa or complex IIb. Complex IIa, consisting of TRADD and Fas-associated protein with death domain (FADD), activates caspase-8 and induces apoptosis through cleavage. Inhibition of RIPK1 ubiquitination results in the induction of complex IIb, composed of RIPK1, FADD and caspase-8. When caspase-8 is inhibited (in either complex IIa or IIb), RIPK1 and RIPK3 form necrosome complexes, activating MLKL through a phosphorylation cascade. Phosphorylated MLKL undergoes oligomerization and migrates to the membrane, which induces necroptosis by membrane rupture or regulating ion flow. [Image was created using Biorender.com (accessed on 16 November 2023)].