Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Martina Derme and Version 2 by Peter Tang.
Prenatal alcohol exposure is responsible for increasing chronic disease risk in later life, including obesity and metabolic syndrome. Alcohol drinking may compromise endogenous antioxidant capacity, causing an increase in free radicals and reactive oxygen species in the newborn. Excessive reactive oxygen species could attack the cellular proteins, lipids, and nucleic acids, leading to cellular dysfunction. Moreover, oxidative stress could play a crucial role in the altered synthesis and release of neurotrophins and progressive mitochondrial modifications with uncontrolled apoptosis.
- fetal alcohol spectrum disorder (FASD)
- prenatal alcohol exposure (PAE)
- oxidative stress
- metabolic disorders
1. Introduction
Prenatal alcohol exposure (PAE) is the foremost avoidable reason for congenital abnormalities and developmental disabilities and affects 2.4–4.8/1000 children [1]. PAE may also raise, in later life, chronic disease risks such as obesity, metabolic syndrome [2], and liver disease [3][4][3,4].
Worldwide, almost 10% of pregnant women drink alcohol. The highest rate of alcoholism during pregnancy is in Europe 25.2%), followed by the American Region (11.2%), the Western Pacific Region (8.6%), the African Region (10.0%), and the South East Asia Region (1.8%). The lowest prevalence is present in the Eastern Mediterranean Region (0.2%) (Figure 1) [5]. Different Mediterranean studies measuring gestational alcohol drinking in women through the analysis of different ethanol metabolites or in the hair, meconium, or urine data showed high variability with values ranging from 3 to 4% up to more than 30% [6][7][8][9][10][11][12][6,7,8,9,10,11,12].
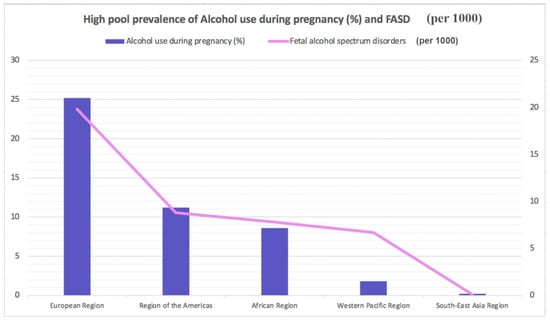
Figure 1.
Highest pooled prevalence of alcohol use during pregnancy (%) and fetal alcohol spectrum disorders (per 1000).
Numerous risk factors have been discovered for alcoholism in pregnancy: older age; higher socioeconomic status, salary, and educational levels; smoking; and unintended pregnancy [6][7][13][14][6,7,13,14].
Fetal alcohol spectrum disorders (FASD) is a “container” word that implies the type of circumstances resulting from PAE. FASD includes disorders such as partial fetal alcohol syndrome (pFAS), fetal alcohol syndrome (FAS), alcohol-related birth defects (ARBD), and alcohol-related neurological developmental disorders (ARND) [15][16][17][18][19][20][21][22][23][24][15,16,17,18,19,20,21,22,23,24]. Several FASD analytic guidelines have been proposed; among the most recent, Hoyme’s guidelines are quite useful [25]. These guidelines were made by a panel of expert authors who analyzed more than 10,000 children with potential FASD. They elaborated a diagnostic process that requires a multidisciplinary approach and a collaboration between pediatricians, geneticists, maternal-fetal specialists, psychiatrists, speech pathologists, physical therapists, audiologists, and ophthalmologists. The advantage of these guidelines is the possibility of elaborating a diagnosis in the prenatal period.
Oxidative stress seems to play a key role in the pathogenesis of both neuropsychiatric and metabolic disorders in pediatrics [26][27][42,43]. Ethanol (EtOH) can alter the endogenous antioxidant ability by depleting the levels of glutathione peroxidase and producing free radicals. Free radicals and reactive oxygen species (ROS), such as hydroxide (HO−) and superoxide (O2−) ions, are derived by O2 partial reduction. They can affect a cell’s structure by damaging nucleic acids, carbohydrates, proteins, and lipids. These molecules are responsible for inducing uninhibited apoptosis of fetal brain damage in children with FASD [28][29][30,44]. The FASD neuropsychiatric effects may be justified by EtOH drinking, inducing the apoptosis of serotoninergic neurons, as shown in rodent models [30][45].
2. Mechanism of Oxidative Stress in Metabolic Disorders
EtOH consumption in pregnancy results in an alteration of oxidative status. A recent case report [31][51] described increased oxidative stress in a mother abusing ethanol drinking during gestation and in her infant a few days after delivery. The FORT (free oxygen radicals test) was used, indeed, to measure the oxidative stress in the mother and her child [32][52]. The FORT is a colorimetric assay based on the ability of transition metals such as iron to catalyze, in the presence of hydroperoxides (ROOH), the formation of free radicals (reactions 1–2), which are then entrapped by an amine derivative, CrNH2. The amine reacts with free radicals, creating a colored, fairly long-lived radical cation, measurable at 505 nm (reaction 3). The color intensity correlates directly to the radical compounds and the hydroperoxide concentrations and, consequently, to the oxidative status of the sample according to the Lambert–Beer law [32][52]. Values superior to 330 U indicate a situation of progressing oxidative stress. Oxidase enzymes (Nox), the mitochondria, and nicotinamide adenine dinucleotide phosphate (NADPH) are the two main apparatuses of ROS production inside the cell [33][53]. The Nox enzymes (Nox1, Nox2, Nox3, Nox4, Nox5, DUOX1, and DUOX2) are cell membrane proteins and Nox2-Nox3 are involved in different pathological circumstances [33][53]. ROS are produced in the mitochondria during oxidative phosphorylation by converting nicotinamide adenine dinucleotide (NADH) to NAD+ [34][35][54,55]. The superoxide anion and Nox2 are quickly converted by the superoxide dismutase enzyme into hydrogen peroxide (H2O2), an important signaling molecule [36][37][56,57]. Indeed, H2O2 is a potent oxidizing agent, and based on these considerations, cells are forced to secrete antioxidant peptides that convert H2O2 to water, including catalase, peroxiredoxin, thioredoxin, and glutathione (GSH) [35][38][55,58]. It is important that H2O2 production is equal to its reduction [39][59]. Pathological diseases such as insulin resistance, obesity, chronic inflammation, hyperglycemia, and dyslipidemia can cause overproduction of ROS [40][41][60,61]. The excessive ROS presence may elicit cellular damage, in particular, peroxidizing lipids and altering DNA [42][62]. Lipid peroxides, lipid peroxidation end products, may be toxic to the cell and should be removed by glutathione throughout a specific mechanism [43][63]. Indeed, previous investigations revealed that patients metabolically affected by the syndrome displayed greater biomarkers of oxidative damage and lower plasma antioxidant enzyme activity than healthy people [44][64]. Peroxidation and nitrosylation can alter nuclear acids and proteins [35][55]. These end products do not typically directly harm the cell [35][55]. However, the increase in inactive proteins may alter the cell’s capability to metabolize them, determining the activation of apoptosis and DNA damage [43][63]. In addition, such elevation in modified proteins reduces their function, leading to severe impairment of regular cell action [36][43][56,63]. The ROS overproduction leads to oxidative stress elevation, which also disrupts redox control and signaling, determining gene expression alteration and increasing stress response elements and growth factors by activating the apoptosis path [39][45][59,65]. Furthermore, oxidative stress may elicit profibrotic and proinflammatory pathways, which alter endothelial dysfunction and insulin metabolic signaling by promoting renal and cardiovascular fibrosis [39][46][59,66].3. Oxidative Stress in Pediatrics after Fetal Alcohol Exposure
FASD is an umbrella expression defining all the circumstances resulting from PAE: partial fetal alcohol syndrome (pFAS), fetal alcohol syndrome (FAS), alcohol-related neurodevelopmental disorder (ARND), and alcohol-related birth defects (ARBD) [47][67]. The vulnerability to ethanol strongly depends on the genetic background of each individual [48][68], and particularly for gestation, it is not possible to establish a consumption-safe level. Indeed, the only practicable recommendation for pregnant women is to avoid alcohol use completely. Damage due to PAE can be long-lasting with no cure [49][69], so early management and correct identification may support prevention and alleviate the metabolic and neurological consequences affecting the FASD person later in life. FASD severity depends on the amount and drinking frequency, as well as the gestational age at which the ethanol was assumed by the pregnant woman [50][51][70,71]. Intervention services, prevention and sensibilization for the mothers could moderate the FASD incidence [52][72]. The fetus has inadequate or null aptitudes in alcohol metabolization and removal [53][73]. Indeed, the several enzymes aimed at ethanol degradation gradually elevate their actions during the various steps of gestation [32][52]. Figure 2 summarizes the mechanisms through which oxidative stress has a major role in the pathogenesis of neurological and metabolic diseases of patients exposed to alcohol in the prenatal period.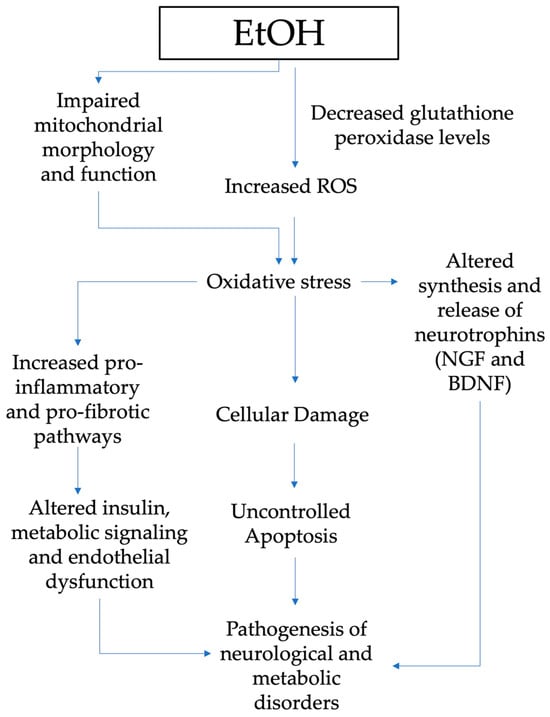
Figure 2. Pathogenesis of neurological and metabolic diseases and role of oxidative stress in patients exposed to alcohol in the prenatal period (EtOH, ethanol; ROS, reactive oxygen species; NGF, nerve growth factor; BDNF, brain-derived neurotrophic factor.