Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Chaiyavat Chaiyasut and Version 2 by Lindsay Dong.
COVID-19 is a global health threat caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and is associated with a significant increase in morbidity and mortality. As a central player in the immune and inflammatory responses, modulating NF-κB activation could offer a strategic avenue for managing SARS-CoV-2 infection.
- COVID-19
- SARS-CoV-2
- NF-κB
- inflammation
- cytokines
1. Introduction
COVID-19, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is a massive health threat all over the globe this century [1][2][1,2], resulting in more morbidities and mortalities. Similarly to the flu virus, COVID-19 is also transmitted through microdroplets when sneezing, coughing, or speaking with the infected person without social distancing. SARS-CoV-2 enters the host through the nasal barrier [3]. The COVID-19 outbreak occurred similarly to pneumonia in December 2019 in Wuhan, China. Soon, the pneumonia-like disease rapidly spread and caused an emergency all over the world. The outbreak was officially announced as a global pandemic in January 2020, and the causative agent is SARS-CoV-2 [4][5][4,5]. The International Committee on Taxonomy named the virus SARS-CoV-2; the disease was named COVID-19 by the World Health Organisation (WHO) [6].
The infection and activation of the virus can result in a cytokine storm through the release of various chemokines and cytokines. Releasing cytokines can cause an inflammatory response that affects the lungs [7]. The COVID-19 outburst is still considered a potent threat to public health. Many research groups started to evaluate the clinical background of COVID-19 infection. The research found that nuclear factor (NF)-kappa B (NF-κB)-driven inflammatory responses [8][9][8,9] are associated with COVID-19. As a result of immune system dysregulation after COVID-19, the unlimited release of proinflammatory cytokines, elevated cytokine levels, and chemokine circulation cause hemorrhage, thrombocytopenia, and systemic inflammation [10].
NF-κB has been an inducible transcriptional model due to its extensive pathophysiological impacts and therapeutical applications [11]. Despite the multifaceted functions of NF-κB, several studies have affirmed that NF-κB operates in conjunction with other signaling pathways and orchestrates diverse responses [12][13][12,13]. Inflammatory cytokines, oxidative stress, and infections can activate NF-kB [13]. The triggered NF-κB engages in diverse cellular signaling pathways that influence cell differentiation, proliferation, survival, intercellular communication, and immunomodulation [14][15][14,15]. Any abnormalities in NF-κB function further lead to inflammatory and autoimmune conditions, metabolic disorders, and cancer [16][17][18][16,17,18].
2. COVID-19 Infection and Pathogenesis
The zoonotic, positive-sense, single-stranded RNA SARS-CoV-2 virus belongs to the family Coronaviridae in the genus Betacoronavirus, subgenus Sarbecovirus [19]. CoV-2 is an enveloped β virus covered with spike glycoproteins and other membrane proteins and showed 80% genetic similarity to CoV-1 and 96.2% to bat CoV RaTG13 [20]. To date, seven human coronaviruses (HCoV) have been found to cause infection in humans, which include alpha coronaviruses (α-CoVs) such as HCov-229E and HCoV-NL63, and beta-coronaviruses (β-CoVs) such as HCoV-HKU1, HCoV-OC43, SARS-CoV, MERS (Middle East respiratory syndrome)-CoV, and SARS-CoV-2 [21]. From observing the past few decades, the epidemics caused by viruses have probably originated from wild animal reservoirs like bats and civet cats, which are then transferred to humans through zoonotic events [22]. In the previous two decennials, three COVID outbreaks have been instigated worldwide. CoVs generally cause lower respiratory tract infection and pathogenicity, resulting in high fatality. SARS-CoV-2 exhibited mild symptoms and severe lung injury and death. HCoV infects humans and causes various clinical conditions, including coughs, moderate febrility, septic shock, progressive respiratory epithelial damage, acute respiratory distress syndrome (ARDS), severe pneumonia, and multiple organ failure in some cases [23][24][23,24]. The genome of the SARS-CoV-2 virus encodes around 16 non-structural proteins (NSP), which are responsible for mass viral replication, and four structural proteins (the envelope, nucleocapsid, membrane, and spike proteins) that produce new virions and other accessory proteins, namely ORF3a, ORF3b, ORF6, ORF7a, ORF8, ORF9b, ORF9c, and ORF10, which are responsible for viral pathogenesis [25][26][27][25,26,27]. Even though the SARS-CoV-2 virus is genetically similar to SARS-CoV-1, several characteristics, such as variabilities in surface proteins and kinetics of viral loads concerning increased transmission rate, differ between SARS-CoV-1 and -2. In SARS-CoV-2 infection, the viral load peaked at the onset of symptoms. It then reduces further, which indicates that CoV2 infectiousness is crucial during the initial first five days after the onset of symptoms [28]. The experiments conducted in vitro on the spike proteins of the virus suggested a potential affinity of the protein to ACE2 [29]. The SARS-CoV-2 virus has a high affinity towards the upper respiratory tract and conjunctiva [30]. SARS-CoV-2 and ACE2 binding results in enhanced transmissibility of viral particles and severity of infection in humans [31][32][31,32]. As with SARS-CoV-2, human CoV-LN63 and SARS-CoV-1 bind to the ACE2 receptor but the severity of the disease differs, which suggests that the potential for pathogenicity of other important virulent factors might differ between these three coronaviruses [33]. In addition to lung injury, extra-pulmonary disturbances were observed in patients [34][37]. SARS-CoV-2 RNA was detected in several other organs, such as the kidney, colon, spleen, skin, heart, and brain [35][36][37][38,39,40], which indicates that those cells might express ACE2 receptors and support the binding of viral particles [35][38][38,41]. The ACE2-RBD of SARS-CoV2 is identical to the RBD of SARS-CoV-1, but SARS-CoV-2 has more specificity and 10 to 20 times higher binding affinity towards ACE2 [39][42]. The intricate configurations of human angiotensin-converting enzyme 2 (hACE2) in conjunction with the RBDs of SARS-CoV-2 variants, namely BA.1.1, BA.2, and BA.3, elucidate that the elevated hACE2 binding affinity exhibited by BA.2 in comparison to BA.1 is attributable to the absence of the G496S mutation in BA.2. Additionally, the R346K mutation observed in BA.1.1 significantly impacts the interaction network within the BA.1.1 RBD/hACE2 interface, inducing long-range alterations. This mutation contributes to the superior hACE2 affinity of the BA.1.1 RBD compared to the BA.1 RBD [40][43]. The ACE2 receptor is the main entry point of the virus, thus allowing it to circulate through the bloodstream and initiate a widespread systemic response characterized by hyperinflammation, which has been linked to various inflammatory diseases [41][44]. HCoV targets the alveolar epithelial cell type II and disrupts the NF-κB pathway, leading to multiple organ failures and fatality [42][46]. Alveolar epithelial type II cells, recognized for their pivotal role, are responsible for synthesizing, secretion, and recycling all constituents of pulmonary surfactant, a critical regulator of alveolar surface tension in mammalian lungs [43][47]. Autopsy studies from COVID-19 patients showed that the pathological changes are similar to those in patients infected with SARS and MERS [44][48]. Pneumonia, lymphopenia, and respiratory failure are common among COVID-19, SARS, and MERS patients [23][45][46][23,49,50]. SARS-CoV-2 infection produces modest respiratory disease accompanied by coughing, fever, headache, diarrhea, dyspnoea, hypoxemia, and severe respiratory failure [19][47][19,51]. Pulmonary vascular leakage, aerated lung tissue loss, and inflammation are the consequences of ARDS. Respiratory failure is accompanied by systemic hyperinflammation, the release of proinflammatory cytokines (IL-1, IL-6, IL-8, and TNF), and other inflammatory markers (Ferritin, C-reactive protein, and D-dimer) [48][52]. The stages of viral infection were explained as viral invasion, replication, disturbed immune response, and multi-organ damage. Once the viral replication is completed inside the target cell, the viral particles are released into the target cells, such as alveolar epithelial cells, and cause damage to them. During this phase, PAMPs (pathogen-associated molecular patterns) and DAMP (damage-associated molecular pattern) molecules are released and induce infiltration of inflammatory cells, cytokines, chemokines, proteases, and free radicals. The cytopathic effects of SARS-CoV-2, reduction of ACE2, imbalance of the renin-angiotensin-aldosterone system, and dysregulated immune response result in a cytokine storm, abnormal blood clotting linked to the release of factors promoting blood coagulation, and microvascular thrombosis triggered by virus-induced blood vessel damage, activation of the complement system, and development of autoimmune reactions [49][50][51][52][55,56,57,58]. The binding of the SARS-CoV-2 spike protein to the ACE2 receptor and the fusion of the viral envelope to the cell membrane mediated by TMPRSS2 induce the first line of the innate immune response. PAMPs are recognized by PRRs (pattern recognition receptors) such as TLRs (Toll-like receptors). Thus, the recognition of these molecules activates transcription factors and IRFs (interferon regulatory factors), resulting in the production of type I interferons (IFNs), chemokines, and proinflammatory cytokines [53][59]. Activation of the immune system recruits inflammatory myeloid cells, neutrophils, CD8 T cells, and NK (natural killer) cells. The cytotoxic function of CD8 T and NK cells helps clear the virus-infected cells [54][60]. Any accumulation of virus-infected cells due to the failed or active clearance of infected cells would produce a hyperinflammatory state called macrophage activation syndrome, ultimately resulting in lung damage [55][56][61,62]. Immune cell profiling of COVID-19 patients’ blood samples indicated that the diminution of CD4+, CD8+, T, and NK cells could cause lymphopenia [1][57][1,63].3. NF-κB Signaling Pathways
Typically, NF-κB remains inactive and confined to the cytoplasm in the quiescent state of most normal cells. It associates with a specific inhibitor known as the IκB protein, which interacts with the Rel homology domain (RHD) of NF-κB, disrupting its nuclear localization sequence functionality. The inhibitor proteins, exemplified by IκBα, IκBβ, and IκBγ, are characterized by the presence of six to seven ankyrin repeats. These ankyrin repeats play a crucial role in facilitating the binding of inhibitor proteins to the RHD of NF-κB, thereby disrupting its nuclear localization sequence functionality [58][65]. The C-terminal shares of the NF-κB2/p100 and NF-κB1/p105 precursors also harbor ankyrin repeats, serving as inhibitors. These IκBs, akin to IκBa, IκBb, and IκBg, play a role in retaining their associated Rel proteins within the cytoplasm. The separation of the NF-κB protein from its inhibitors is necessary to activate NF-κB. Two primary signaling pathways, canonical and non-canonical, govern the dissociation of IκB protein inhibitors from the NF-κB dimer [59][66].3.1. Canonical NF-κB Pathway
The canonical NF-κB pathway governing NF-κB activation is dependent upon the inducible degradation of IκBs, notably IκBα. It concludes in the nuclear translocation of diverse NF-κB complexes, with a predominant occurrence of the P50/RelA dimer [60][61][68,69]. The inducible degradation of IκBα is carried out through its phosphorylation, a process catalyzed by the IκB kinase (IKK). The IKK is a trimeric complex consisting of two catalytic subunits, namely IKKα and IKKβ, alongside a regulatory subunit identified as IKKγ, also referred to as the NF-κB essential modulator (NEMO) [62][70]. In canonical NF-κB signaling cascades, such as those initiated downstream of tumor necrosis factor receptor 1 (TNFR1), IKKβ is essential and adequate for the phosphorylation of IκBα at Ser32 and Ser36, as well as IκBβ at Ser19 and Ser23. IKKα can facilitate IκBα phosphorylation, playing a pivotal role in canonical NF-κB-dependent transcriptional responses. Despite some exceptions, wherein specific instances are detailed below, it is observed that both canonical and non-canonical pathways employ TNF receptor-associated factor (TRAF) family members for activation. Notably, in the canonical pathway, NEMO-dependent signaling leading to typical IκBs, along with the involvement of receptor-interacting proteins (RIPs), is also required [60][68]. RIPs serve as pivotal adapters in canonical NF-κB signaling. These proteins exhibit dual functionality by acting both upstream of and accompanied by TRAF proteins to activate IKK. In the NF-κB signaling pathways, RIPs function as authentic adapters by engaging with upstream signaling modules through well-characterized protein-binding domains. Moreover, RIP family members apply significance without a clear requirement for TRAFs. These pathways, characterized by RIP-dependent and TRAF-independent IKKβ activation, may encompass antigen receptor signaling and responses to DNA damage [60][68]. Signaling to IKK downstream of RIPs and TRAFs involves several kinases implicated in NF-κB signaling pathways. Specifically, in canonical NF-κB pathways, this function is predominantly executed by TGFβ-activated kinase-1 (TAK1) [63][64][71,72].3.2. Non-Canonical NF-κB Pathway
The non-canonical NF-κB signaling pathway is responsible for the activation of the p52/RelB NF-κB complex through a distinctive mechanism predicated upon the inducible processing of p100 instead of the conventional degradation of IκBα [65][67]. Beyond its role as the precursor of p52, p100 serves a dual function similar to an IκB-like molecule, applying a preferential inhibitory effect on the nuclear translocation of RelB [66][73]. At the core of the non-canonical pathway lies NF-κB-inducing kinase (NIK), a pivotal signaling component that assimilates signals from a subset of TNF receptor family members. NIK, in turn, initiates the activation of IκB kinase-α (IKKα), a downstream kinase responsible for initiating the phosphorylation and subsequent processing of p100 [65][67]. NIK is the initial element discovered in the non-canonical NF-κB signaling pathway [67][74]. NIK, categorized as a MAP kinase kinase kinase (MAP3K), was initially implicated in the activation of NF-κB through the TNF receptor (TNFR) pathway [68][75]. NIK applies its functional role by activating a subsequent kinase, IKKα [69][76]. The recruitment of NIK by receptors has the potential to elevate its local concentration, thereby potentially inducing autophosphorylation without necessitating an increase in NIK expression. This mechanism of NIK activation may be operative during the initial stages of non-canonical NF-κB signaling, suggesting a probable scenario wherein NIK accumulation could play a role in sustaining non-canonical NF-κB signaling. Nevertheless, an alternative perspective suggests that the activation of NIK by specific receptor signals, especially within distinct cell types, may exclusively rely upon the early-phase mechanism [65][67]. Genetic evidence indicates that this NF-κB pathway governs critical biological processes, including lymphoid organogenesis, the survival and maturation of B cells, activation of dendritic cells, and the regulation of bone metabolism [70][78].4. NF-κB Signaling in COVID-19 Infection
Generally, the NF-κB transcription factor regulates immune cell functions and gene expression to various pathogenic stimuli. Thus, the proinflammatory stimuli induce the NF-κB signaling cascade. In the case of COVID-19 infection, the abnormal NF-κB activation results in elevated levels of immune cells and cytokine storm. The activation and strong involvement of NF-κB during COVID-19 infection show that NF-κB might be a potential target [71][81]. COVID-19 patients displayed elevated levels of IL-1β, IL-6, and TNF [72][73][82,83]. Such an enhanced cytokine storm induces ARDS and high mortality rates among COVID-19 patients [74][84]. The higher secretion of cytokines can also enhance other viral infections, including hantavirus pulmonary syndrome, H5N1, influenza virus, and SARS-CoV-1 [75][76][77][78][85,86,87,88], and produce a great impact over the negative feedback control of the immune response, resulting in the continuous cytokine secretion that perpetuates the positive feedback response of immune cells. Recruiting even more immune cells towards the infection site causes enhanced inflammation, COVID-19 disease progression, multiple organ failures, and fatality [79][89]. The function of NF-κB is modulated by an IκB (inhibitor of NF-κB) family of proteins such as IκBα, β, γ, and ε, and Bcl-3 [80][81][90,91]. Highly conserved NF-κB dimeric transcription factors include Rel-like domain-containing proteins RelA, RelB, RelC, p105, and p100. P105 and p100 are latent forms of NF-κB proteolytically cleaved to generate active forms, namely p50 and p52 [80][90]. RelA and p50 form heterodimers and act as major NF-κB complexes in most cells. Studies have shown that NF-κB regulates 400 human genes that code for cytokines, chemokines, and genes responsible for stress response, cell growth, and cell death [82][92]. Basal expression of NF-κB is observed in cells under pathological circumstances, including exposure to pathogenic stimuli, stress hormones, radiation, and free radicals [83][93]. The phosphorylation and subsequent degradation of IκBβ result in the release of p50 and p52 subunits, which then translocate into the nucleus. Within the nucleus, these subunits serve as putative transcription factors, governing the transcription of genes essential for the inflammatory response [84][85][94,95]. Viral infections trigger the host’s innate immune system, which recognizes the viral particles and products, including nucleic acids, capsids, and other proteins. It induces a defense mechanism by initiating an NF-κB signal, attenuating viral infection [82][92]. The upregulation of NF-κB-mediated inflammasomes (receptors/sensors of the innate immune system) modulating the activation of caspase-1 and responsible for eliciting an inflammatory response in reaction to infectious microorganisms and molecules originating from host proteins [86][100] following viral invasion and infection has been documented [87][101]. Viruses evolved with different infection strategies, including targeting ubiquitination to modulate NF-κB signaling by evading host surveillance. The viral genome encoding a viral E3 ligase targets components of the NF-κB proteasome, inducing proteasomal degradation and consequently suppressing NF-κB signaling. Few viruses encode a viral deubiquitinating enzyme to remove the K63-linked polyubiquitin chain and prevent NF-κB activation. In the case of SARS-CoV, the papain-like protease relieves the K63-linked polyubiquitin chain of TRAF3 and TRAF6 and blocks the activation of interferon-regulatory factor-3 and NF-κB [88][89][90][106,107,108] (Figure 13).
Figure 13. SARS-CoV-2 papain-like protease inhibits the TLR3 and TLR7 signaling pathway-mediated inflammatory and antiviral response. Papain-like protease decreases the Lysine 63 (K63)-linked polyubiquitin chains of TRAF3 and TRAF6, which leads to reduced E3 ubiquitin ligase activity from TRAF3 and TRAF6, thereby preventing the activation of IRF-3 and NF-κB and further inhibiting TLR3- and TLR7-mediated expression of INF1 and proinflammatory cytokines. TLR3: Toll-like receptor 3; TLR7: Toll-like receptor 7; NF-κB: Nuclear factor kappa B; IRF-3: Interferon regulatory transcription factor 3; TRAF3: tumor necrosis factor receptor (TNF-R)-associated factor 3; TRAF6: tumor necrosis factor receptor (TNF-R)-associated factor 6; INF1: Interferon 1.
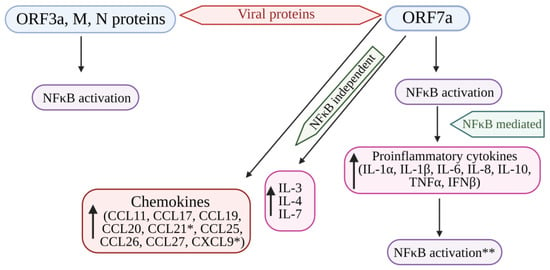
Figure 24. Cytokine storm induced by SARS-CoV-2 proteins (ORF7a, ORF3A, M, and N proteins) mediated by the activation of NF-kB. The structural viral proteins (M and N proteins) and predicted accessory viral proteins (ORF3a and ORF7a proteins) activate the NFκB signaling pathway. Among the 4 viral proteins, ORF7a is the most potent activator of proinflammatory cytokines, whose expression is mediated by NFκB activation. Cytokines such as IL-3, IL-4, and IL-7 were also upregulated by ORF7a. In addition, ORF7a promotes the expression of chemokines. The over-expression of cytokines further activates the NFκB pathway, resulting in the SARS-CoV-induced cytokine storm. * Significantly elevated in COVID-19 patients. ** Overexpression of cytokines further enhances NFκB activation. NFκB: Nuclear factor kappa B; ORF7a: Open-reading frame 7a; ORF3a: Open-reading frame 3a; M: Membrane protein; N: Nucleocapsid protein; IL: Interleukin; TNFα: Tumor necrosis factor-α; IFNβ: Interferon-β; CCL11, 17, 19, 20, 21, 25, 26, 27: Eosinophil chemotactic factors; CXCL9: Chemokine; ↑: Increased or upregulated.
Viruses activate the NF-kB signaling cascade to promote infection; viruses interact with NF-κB signaling components, especially PRRs and IκB kinase [84][92][94,110]. Certain viruses, including herpes simplex virus-1, human immunodeficiency virus, and bovine foamy virus, promote infection and viral gene expression using NF-κB transcription factors [92][93][94][95][110,111,112,113]. SARS-CoV-2 infection induces an imbalance in the inflammatory responses through weak production of IFNs and hyperproduction of proinflammatory cytokines, leading to ARDS [96][97][98,114]. Several clinical studies have postulated that cytokines (IL-1, IL-6, IL-8, and TNF-α) and chemokines were abnormally raised to high levels in COVID-19 patients [23][72][98][99][23,82,115,116]. NF-κB governs the manifestation of proinflammatory cytokines, and through a positive feedback mechanism, these cytokines further enhance NF-κB activity [100][117]. In COVID-19 patients, increased levels of proinflammatory cytokines intensify NF-κB activation [98][115]. The upregulation of NF-κB likely promotes the inflammatory response in COVID-19 patients. The viral proteins responsible for immune activation and the exact molecular mechanisms behind such activation have remained unidentified.
SARS-CoV-2 induces a substantial increase in proinflammatory cytokine production (Figure 24). Previous research on SARS-CoV-1’s ORF3a, M, ORF7a, and N proteins demonstrated the boost in NF-κB activity [101][102][103][103,118,119]. The papain-like protease plays a crucial role in viral replication by cleaving a specific site within the viral replicase and eliminating ubiquitin from cellular proteins. In SARS-CoV, the papain-like protease removes the Lys63-linked ubiquitin chains from TRAF3 and TRAF6 [88][106]. Papain-like protease disrupts the polyubiquitination process of IκBα and prevents the activation of NF-κB [104][120] (Figure 13). Persistent activation of NF-κB in individuals with established metabolic syndromes elevates their susceptibility to cardiac complications; with coupled cytokine activation, this condition can result in cardiac injury [105][64].
5. NF-κB Pathway as a Pharmacological Target in COVID-19
5.1. Inflammatory Response Triggered by the SARS-CoV Spike Protein
The study found that immune cells (macrophages) release the inflammatory proteins (IL-6 and TNF-α) while exposed to the spike protein of SARS-CoV. The release of IL-6 and TNFα depends on the spike protein quantity and exposure duration, with the breakdown of IκBα playing a crucial role in NF-κB activation. Inhibiting this step reduces inflammatory protein release, highlighting NF-κB as a key signaling pathway for the spike protein-induced inflammatory response in SARS-CoV [106][135].
Drugs constraining NF-κB activation, such as caffeic acid phenethyl ester, Bay 11–7082, and parthenolide, lowered inflammation by reducing the production of inflammatory molecules in mice infected with SARS-CoV. Furthermore, blocking NF-κB helped protect against lung damage and improved the survival of the mice after SARS-CoV infection [107][104]. Research reported that during the SARS-CoV and MERS-CoV outbreaks, viral proteins such as NSP1, NSP3a, NSP7a, spike proteins, and nucleocapsid proteins activated the NF-κB pathway, thereby increasing disease severity and fatality [80][101][107][90,103,104].