Plants are an untapped natural resource; their secondary metabolites take part in a variety of pharmacological activities, making them an essential ingredient in the synthesis of novel medications and the source of reserve resources in this process. Hepatitis and liver cancer are two conditions that can result from non-alcoholic fatty liver disease (NAFLD). NAFLD is a condition that now affects a significant section of the global population. There is a need for preventative action on predisposing factors. Due to their effectiveness and few side effects, herbal medications are frequently utilized for the prevention and treatment of NAFLD.
- non-alcoholic fatty liver disease (NAFLD)
- polyphenols
- plants
- natural products
- anti-steatotic compounds
1. Introduction
2. Pathophysiology of NAFLD
Fatty liver disease includes two distinct categories: alcoholic fatty liver disease (AFLD) and non-alcoholic fatty liver disease (NAFLD) [15]. The first is triggered by excessive alcohol consumption; the underlying causes of the second condition have not yet been fully clarified, but a multifactorial etiology is clearly emerging. It recognizes various elements, including obesity, associated insulin resistance (IR), type 2 diabetes mellitus, dyslipidemia, atherosclerosis, hypertension, and fatty liver disease itself [16][17][16,17]. Interestingly, these are the same risk factors associated with cardiovascular disease (CVD). In patients with NAFLD, liver enzymes have been shown to predict the incidence of CVD, independent of traditional risk factors, including C-reactive protein and metabolic syndrome (MS). Indeed, the extent of liver damage has been correlated with early carotid atherosclerosis, suggesting that both vessel and liver damage share similar inflammatory mediators [18]. For these reasons, NAFLD has recently been proposed as an early marker of atherosclerosis and endothelial dysfunction and, consequently, as a cardiovascular risk factor. Considering the wide spectrum of effects often related to NAFLD, several scientists have recently provided a more accurate terminology for this disease. The term NAFLD has been changed to “MAFLD,” meaning metabolic dysfunction associated with fatty liver disease. Therefore, NAFLD can be regarded as the hepatic manifestation of a metabolic syndrome [19][20][21][19,20,21]. In this MS, also the intestinal microbiota plays a key role. Recent studies have shown that, in addition to genetic predisposition and diet, the intestinal microbiota influences the hepatic metabolism of carbohydrates and lipids [22]. It also appears to affect the balance between pro-inflammatory and anti-inflammatory effectors in the liver, thereby affecting the progression of MAFLD [23]. Changes in the composition of the intestinal microbiota, in fact, increase intestinal permeability and, consequently, the translocation of bacteria and their products; this induces endotoxemia, which contributes to liver inflammation in patients with NASH [24]. Species belonging to the Gram-negative class grow during MAFLD, while those belonging to the Gram-positive class decrease [25][26][25,26]. Therefore, the gut microbiota may be a useful tool as a predictor for the progression and severity of MAFLD [27][28][27,28]. Among the first hypotheses about etiopathogenesis, we have that of Day et al., 1998 [29]. The authors proposed the so-called “two-hit” pathogenetic model to explain the appearance of NAFLD and its progression to NASH. The first “hit” is the accumulation of triglycerides in the liver (steatosis) due to several conditions. The liver may become overloaded by excessive dietary intake, increased TG synthesis, excessive influx of fatty acids into the liver as a result of lipolysis of adipose tissue, decreased export of lipids from the liver in the form of VLDL, or decreased oxidation of fatty acids. In this model, the first hit must be followed by a subsequent insult (second hit), capable of activating a consistent lipid peroxidation that generates reactive oxygen species (ROS) and cytotoxic aldehydes that trigger processes such as the transcription of NF-kB (regulator of inflammation genes), leading to necro-inflammation and fibrosis [29]. It has recently emerged that free fatty acids have a direct ability to promote liver injury and the activation of inflammation, even in the absence of a second hit [30]. The initial two-hit theory became obsolete and was reformulated in the form of a “multi-hit” hypothesis. The “hits” could be represented by several injuries, such as obesity, insulin resistance, oxidative stress, and pro-inflammatory processes [31][32][31,32]. Tilg and Moschen, 2010 [33] revised this pathogenetic reconstruction, considering NASH as distinct from simple fatty liver disease and not an evolution of the latter. The authors have proposed this new model in which multiple causal factors, acting simultaneously, are able to determine the onset of liver disease (the “parallel multiple stroke model”) [33]. According to this interpretation, steatohepatitis and steatosis represent two distinct nosological entities. Inflammation may also precede and lead to the onset of steatosis in NASH. In fact, there is evidence of patients with NASH who have almost no steatosis and of regression of steatosis in mice models with the obese phenotype ob/ob after anti-inflammatory therapy with anti-TNF-α monoclonal antibodies or metformin (also capable of reducing the expression of TNF-α). The hypothesis of Tilg and Moshen, 2010 [33] brings to light the prominent role of factors of extrahepatic derivation in determining inflammation and secondary liver fibrosis [33]. Genetic factors, such as polymorphisms of some phospholipases, modifications of the intestinal microflora (dysbiosis)—secondary to a diet rich in fats/carbohydrates—insulin resistance, an increase in circulation of bacterial endotoxins (in relation to an altered liver sequestration function), reduced synthesis of short-chain fatty acids (capable of performing an anti-inflammatory action), and an altered expression in the intestinal epithelium of toll-like receptors (TLRs) (regulators of inflammation and involved in innate immunity), may represent “molecular mediators” of liver damage, inducing lipid accumulation in the liver, lipotoxicity, fibrosis and, finally, the onset of neoplasms. In the next section, the causes that contribute to the onset of liver disease are analyzed.Accumulation of Lipids and Steatosis
The control of lipid metabolism involves many critical organs, including the liver. The buildup of TG in hepatocytes is a hallmark of NAFLD. It is believed that there is an imbalance between the amount of FA input (uptake and synthesis with subsequent esterification to TGs) and outflow (oxidation and secretion), which results in hepatic steatosis. Hepatic fatty acids, used for TG synthesis, derive from diet, lipolysis of adipose tissue, or de novo lipogenesis in the presence of excess glucose [1]. According to research by Donnelly et al., 2005 [34], approximately 59% of liver-free fatty acids (FFAs) in NAFLD patients come from circulation, 26% from de novo lipogenesis, and 15% from food [34]. From the small intestine, dietary fatty acids are absorbed, organized into lipoprotein-rich particles (chylomicrons), and released into the blood and lymph. The liver receives the remaining chylomicrons, with the majority going to adipose tissue. The liver cells will not be able to get rid of the extra lipid if the fat meal is repeated often; if the exposure is just once in a while, the triglyceride elimination is accomplished quickly. Increased lipid mobilization from adipose deposits has the same result as intracellular triglyceride buildup. [35]. Typical steatosis of this kind occurs in diabetics, in which lipolysis is activated, and lipemia remains high due to insulin deficiency [36]. There are instances when lipid overflow develops inside the cell owing to increased fatty acid synthesis; this circumstance is known as de novo lipogenesis. The transcription factors SREBP-1c (sterol regulatory element binding protein-1c) and ChREBP (carbohydrate-responsive element-binding protein), which regulate the key lipogenesis genes, ensure that acetyl-CoA is converted to fatty acids in the liver. Insulin is primarily responsible for this conversion [37]. In patients with NAFLD, it is common to find an overexpression of the genes involved in the de novo synthesis of fatty acids, such as acetyl-CoA carboxylase 1 and 2 (ACC1, ACC2) and fatty acid synthase (FAS) [35]. Therefore, insulin plays a role in NAFLD appearance. Specific receptors found on the plasma membrane of the cells allow insulin to carry out its activity. When blood glucose levels rise after a meal, pancreatic beta-cells naturally generate insulin to lower them. Several target proteins are activated, and a cascade of cascading processes is set off when insulin binds to the cell receptor. Insulin is also responsible for de novo lipogenesis in the liver, as previously mentioned. Another function attributable to insulin is the inhibition of lipolysis in adipose tissue. NAFLD often develops in those who have insulin resistance. Increased consumption of free fatty acids (FFAs), hyperglycemia, which promotes the formation of free radicals (glycotoxicity), and inflammatory cytokines released by adipocytes are the key contributors to insulin resistance [38]. In the case of IR, the adipose tissue’s resistance to insulin triggers lipolysis, which increases the release of free fatty acids from the adipose tissue. These free fatty acids are then transported to the liver via the hepatic artery and portal vein, increasing the influence of hepatic fatty acids [39]. High blood glucose levels (in the context of IR) lead to hyperinsulinemia, which activates SREBP-1c and encourages de novo hepatic lipogenesis (through the malonyl- and acetyl-CoA pathways). The increased lipogenesis is also indirect due to the increased production of malonyl-CoA, which inhibits carnitine-palmitoyl transferase-1 and the entry of fatty acids into the mitochondria by reducing ꞵ-oxidation, thus increasing the accumulation of fatty acids and triglycerides [40] (Figure 1).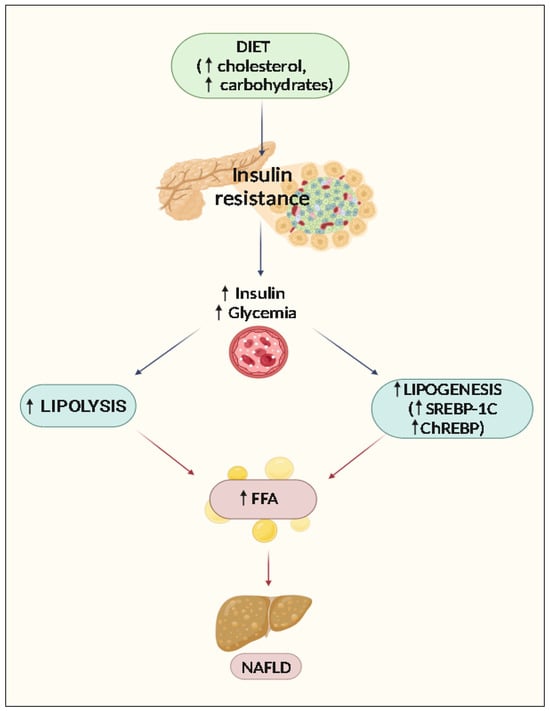
3. Progression of Liver Damage: The Role of Oxidative Stress, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress
It has been shown that individuals with NASH have biochemical and ultrastructural lesions of the mitochondria; these lesions are not completely understood at this time, but it is thought that TNF, non-esterified fatty acids and lipoperoxidation products may be to blame individually or in combination. Non-esterified fatty acids uncouple oxidative phosphorylations in this way. This condition, which is typically accompanied by an increase in oxygen consumption, causes an increase in the production of ROS, which in turn lowers the concentration of antioxidants, causes lipoperoxidation, and mediates the release of TNF from Kupffer cells (KC), which are inflammatory cytokines that are also released by steatotic hepatocytes and increase their impact by activating Kupffer cells. ROS has a growing role in the activation of Ito cells (hepatic stellate cells (HSC)), which leads to hepatic fibrogenesis, a late effect of NASH. Liver steatosis is mostly linked to oxidative stresses, mitochondrial dysfunction, and endoplasmic reticulum (ER) stress [41] (Figure 2).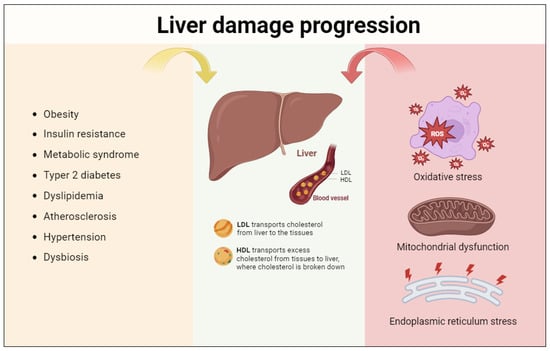
3.1. Oxidative Stress
As it was feasible to infer, a major mechanism underlying NASH is oxidative stress (OS), which is defined as an imbalance between oxidant and antioxidant chemical species. ROS are hazardous chemicals that the body’s antioxidant system must eliminate through interacting with proteins, lipids, and nucleic acids [42]. Oxidative stress is brought on by either increased ROS production in liver cells or weakened antioxidant defenses. The body’s fight against free radicals is made up of both enzymatic and nonenzymatic processes. While glutathione (GSH), ascorbic acid (vitamin C), retinol (vitamin A), and tocopherol (vitamin E) are nonenzymatic systems that can protect biomolecules and cellular structures from ROS damage by acting as electron receptors, superoxide dismutase (SOD), catalase (CAT), and GPx are examples of enzyme systems. In the context of the liver, hepatocytes counteract the harmful effects of ROS to avoid cellular damage by turning on antioxidant enzymes such as heme oxygenase 1 (HO1), NAD(P)H:Quinone Oxidoreductase 1 (NQO1), and SOD. It is significant because many of the genes responsible for encoding these antioxidant enzymes are controlled by the transcription factor nuclear factor-erythroid 2-related factor 2 (Nrf2) [43]. Nrf2 protects against NAFLD in addition to activating genes that encourage the removal of ROS by negatively regulating genes that encourage lipid synthesis [44]. Damage to cell membranes and modifications to lipid, protein, carbohydrate, and DNA biomolecules are brought on by ROS [45]. OS is generated in mitochondria, peroxisomes, and ER, organelles in which oxygen consumption is high [46]. Lipid peroxidation, which causes the histological characteristics seen with NASH, is the primary modification mediated by reactive oxygen species [47]. By uncoupling the oxidative cycle, FFA promotes the expression of the microsomal monooxygenases CYP4A and CYP2E1, which are in charge of producing ROS [45]. CYP2E1 is mainly expressed in the liver, with the highest expression in hepatocytes. The ER is where CYP2E1 is mostly found, although the mitochondria also express it. A wide range of compounds, including numerous drugs, polyunsaturated fatty acids, ethanol, paracetamol, and the majority of organic solvents, are metabolized by CYP2E1. The expression of CYP2E1 mRNA and protein is regulated by a number of substances, including insulin, acetone, leptin, adiponectin, and cytokines. Because insulin inhibits the cytochrome CYP2E1, peripheral insulin resistance results in increased levels of its expression [48]. According to Chen et al., 2020, [49], OS is linked to the release of pro-inflammatory cytokines, lipid peroxidation, and disruption of phospholipid membranes, all of which cause hepatocellular damage and subsequently affect the course of NAFLD [32]. HSC, the primary cells responsible for the extracellular fibrous matrix deposition that is intimately related to liver fibrosis, are likewise activated by ROS [50].3.2. Mitochondrial Dysfunction
Fat acid oxidation process imbalances may contribute to an excessive buildup of lipids in the liver. This process is normally proportional to the concentrations of fatty acids released by the adipose tissue following the stimulus provided by glucagon. Oxidation takes place in the three main subcellular organelles of the organism: the mitochondrion and the peroxisomes, which carry out β-oxidation, and the ER, which carries out the ω-oxidation, catalyzed by cytochrome CYP4A. Several cytochrome P450 enzymes that may hydroxylate the omega terminal carbon and, to a lesser degree, the omega 1 position of saturated and unsaturated fatty acids, as well as enzymes involved in the omega hydroxylation of different prostaglandins, are encoded by the CYP4A subfamily [51][52][51,52]. Peroxisome proliferator-activated receptor-α (PPARα), a nuclear hormone receptor that controls the oxidation and transport of fatty acids, controls the activity of some of the main enzymes in these three oxidation systems [34]. As soon as it is activated, it forms a heterodimer with the retinoid X receptor (RXR) and binds to peroxisome response elements in the genes responsible for fatty acid oxidation. According to Suhaili et al., 2017 [53], lipotoxicity and increased inward flux of FFA cause lysosomal membrane permeabilization and mitochondrial dysfunction, which in turn trigger hepatocyte apoptosis. Hepatocyte apoptosis causes an immune response that generates a release of proinflammatory cytokines; this leads to further aggravation of liver inflammation [53]. Mitochondrial β-oxidation is implicated primarily in the oxidation of short (<C8), medium (C8–C12), and long (C12–C20) chain fatty acids. As a result of this process, fatty acids gradually shorten into minor units of acetyl-CoA, which can either be condensed to create ketone bodies (oxidizable substrates that serve as an alternative source of energy for extrahepatic tissues) or enter the tricarboxylic acid cycle to further oxidize into water (H2O) and carbon dioxide (CO2). Carnitine palmitoyl-transferase 1a (CPT1a), which is in charge of moving fatty acids from the cytosol to the mitochondrion, the level of carnitine, and the substrate malonyl-CoA, which inhibits CPT1a, are the primary regulators of β-oxidation [54]. Four different dehydrogenases, each particular for the length of the fatty acid chains, catalyze the initial stage of mitochondrial β-oxidation. A single trifunctional mitochondrial protein (MTP), which catalyzes three distinct enzymatic processes (2-enoyl-CoA hydratase, 3-hydroxyacyl-CoA dehydrogenase, and 3-ketoacyl-CoA thiolase), is responsible for the second, third, and fourth stages. Long-chain fatty acids (>C20) undergo a shortening process in peroxisomes before being transported to the mitochondrion, where they carry out the β-oxidation process. The long-chain dicarboxylic acids (highly toxic to the mitochondrion), generated by microsomal ω-oxidation, are metabolized by the peroxisomal β-oxidation system. Although ω-oxidation is the least important metabolization pathway, in conditions of fatty acid overload, such as obesity or diabetes, large amounts of dicarboxylic acids are formed, inducing greater activation of this metabolic pathway [55]. CPT1a expression is 50% lower in NAFLD patients, although long-chain acyl-CoA dehydrogenase (LCAD) and long-chain L-3-hydroxyacyl-CoA dehydrogenase-α (HADHα) are more expressed. Uncoupling protein 2 (UCP2), which regulates the loss of protons along the inner membrane and uncouples the oxidation energy from ATP synthesis, is a significant enzyme involved in the mitochondrial regulation of ROS. NAFLD patients have increased levels of this enzyme [55][56][55,56]. Mitochondrial damage and the increase of aberrant mitochondria in the liver generate excessive oxidative stress responsible for advancing the NAFLD course [57].3.3. Endoplasmic Reticulum Stress
The ER is the organelle in which secretory and membrane proteins achieve correct folding, thanks to the presence of chaperones. The pathogenesis of NASH is influenced by variables including stress (short of nutrition and viral infections), increased production of misfolded proteins, and ER stress, which promotes the activation of a number of transcription factors and kinases [58]. Through the SREBP pathway, the activated transcription factors cause an increase in lipid production, as well as the transcription of proapoptotic genes. The activation of ER kinases, such as inositol-requiring enzyme 1α (IRE1α) and PKR-like ER kinase (PERK), in turn, involves the induction of stress kinases, such as c-Jun N-terminal kinase 1 (JNK1), which may contribute to the induction of apoptotic phenomena [59].4. Natural Compounds Showing Activity towards NAFLD
Numerous studies have demonstrated the potential of antioxidants from natural sources, including foods and plant species, to prevent or treat NAFLD. Over the years, research has been able to identify alkaloids and flavonoids in addition to polyphenols. The biological effects of the flavonoid, phenolic, alkaloid, and terpene constituents are shown in Figure 3.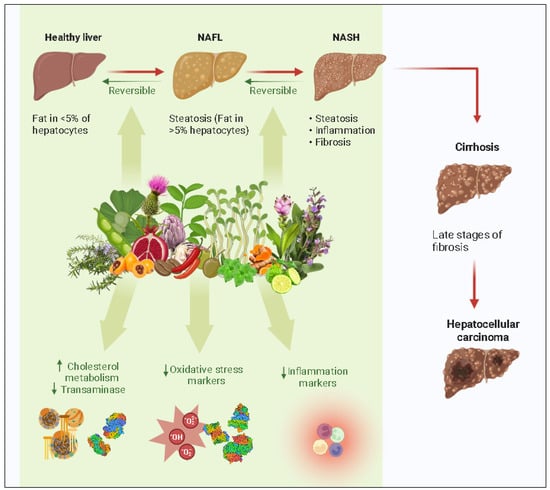
|
Flavonoids Component |
Effects |
References |
|---|---|---|
↑: Increase, ↓: decrease of expression, concentrations, or activity. ACC: acetyl-CoA carboxylase; AKT: serine/threonine kinase 1; ALT: alanine aminotransferase; AMPK: adenosine monophosphate-activated protein kinase; ARE: antioxidant responsive element; AST: aspartate aminotransferase; CAT: catalase; C/EBP: CCAAT/enhancer binding protein; CPT-1a: carnitine palmitoyl transferase-1; FAS: fatty acid synthase; SCD-1: stearoyl-CoA desaturase 1; FFA: free fatty acids; FOXO-1: forkhead box protein O1; GCK: germinal center kinase; GPx: glutathione peroxidase; GSH: glutathione; HO-1: heme oxygenase-1; iNOS: inducible nitric oxide synthase; IL: interleukin; IRE1a: inositol-requiring enzyme 1a; LDL: low-density lipoprotein; LKB1: liver kinase B1 (STK11); MAPK: mitogen-activated protein kinase; MDA: malondialdehyde; MPO: myeloperoxidase; MTTP: microsomal triglyceride transfer protein; MyD88: myeloid differentiation primary response gene 88; Nrf2: Nuclear factor erythroid 2-related factor 2; PI3K: phosphoinositide-3-kinase; PPARα: peroxisome proliferator-activated receptor α; RBP4: retinol-binding protein-4; ROS: reactive oxygen species; SIRT1: silent mating type information regulation 2 homolog 1; SOD: superoxide dismutase; SREBP-1c: sterol regulatory element binding protein-1c; TC: total cholesterol; TLR4: Toll-like receptor 4; TG: triglycerides; TNF: tumor necrosis factor; UCP1: uncoupling protein-1; VLDL: very low density lipoprotein; WAT: white adipose tissue; and XBP1: X-box-binding protein 1.
|
Plants |
Effects |
References |
|||
|---|---|---|---|---|---|
Quercetin |
|||||
|
Tea (Camellia sinensis) |
↓VLDL, ↓LDL, ↓TC, ↓TG, ↓FFA, ↓ALT, ↓AST, ↓PI3K/AKT pathway, |
↓ALT, ↓AST, ↓NADPH, ↓MPO, ↓RO, ↓Lipid peroxidation ↑IRE1a/XBP1 pathway, ↓NAFLD activity score, ↓Lipid peroxidation, ↓TNFα, ↓IL-6, ↑SIRT1, ↓NF-kB, ↓iNOS, ↓TLR4 |
[57][ |
[102]58][103][59][104[60]][57,58[,59]61],60[62],61[,11163,112] |
|
[ |
Anthocyanins |
||||
|
Curcuma (Curcuma longa) |
↓TC, ↓TG, ↓LDL, ↑HDL, ↓ALT, ↑PPARα, ↓TLR4, ↓MAPK pathway, ↑AMPK pathway, ↓MPO, ↓MCP, ↓Fibrosis score, ↓WAT, ↓TNFα, ↓IL-1β, ↓IL-6, ↑IL-10, ↑IL-22, ↓ROS, ↑SOD, ↓COX-2, ↓iNOS |
↓TC, ↓TG, ↓LDL, ↑HDL, ↓ALT, ↓AST, ↓ROS, ↓Lipid peroxidation, ↓Cyt-c, ↓CASP 3, ↓CASP 8, ↓NF-kB, ↑AMPK, ↓SREBP-1c, ↓FAS, |
|||
[ |
Kaempferol |
↓TC, ↓ALT, ↓AST, ↑GSH, ↓MDA, ↑SIRT1, ↑CPT-1a, ↓SREBP-1c, ↓FAS, ↓SCD-1, ↓PPAR, ↓C/EBP, ↑SOD, ↓NF-kB ↓PARP1, ↓FOXO-1 |
|||
, | |||||
|
Loquat (Eriobotrya japonica) |
↓ALT, ↓AST, ↓ROS, ↓TGF, ↓Collagen |
Hesperidin |
|||
|
Ginkgo (Ginkgo biloba) |
↓TC, ↓ALT, ↓AST, ↓RBP4, ↓H-FABP, ↓ROS, ↑HO-1, ↑PI3K/AKT-Nrf2 pathway, ↑SIRT1-AMPK pathway, ↑SOD, ↑GPx, ↑GSH, ↓TNFα, ↓IL-1β, ↓IL-2, ↓IL-6, ↓NF-kB |
][ |
↓TC, ↓LDL, ↓TG, ↑HDL, ↓ALT, ↓AST, ↑CPT-1a, ↓TNFα |
||
[ |
Baicalein |
↓TC, ↓ALT, ↓AST, ↓LDL, ↓TG, ↓FFA, ↓TNFα, ↓SREBP-1c, ↓SREBP-1c, ↓TG, ↓GLU, ↓FAS |
|||
|
Olive tree (Olea europaea) |
↓TRX-1, ↓4-HNE, ↓ROS, ↑Nrf2, ↓SOD, ↓CAT, ↓GPx, ↓IL-6, ↓IL-8, ↓TNFα |
||||
[ |
Troxerutin |
||||
|
Pomegranate (Punica granatum) |
↑CYP1A1, ↑PGC-1α, ↑CPT-1 β, ↑GSH, ↑PPARα, ↓SREBP-1c, ↓ROS, ↓NF-kB, ↓FATP-1, ↓SCD1 |
↓TC, ↓TG, ↓LDL, ↑PPAR, ↑CPT-1a, ↑ACO, ↓SCD-1, ↓ALT, ↓AST |
|||
[ |
Phenolic components |
||||
|
Milk thistle (Silybum marianum) |
↑AMPK activity, ↓PPAR-γ, ↓ACC, ↓ROS, ↓FAS, ↓SIRT1, ↑CAT, ↑NAD+ homeostasis, ↑IRS-1/PI3K/Akt pathway, ↓FXR, ↓NF-kB, ↓4-HNE, ↓glutathione depletion, ↓mitochondrial hydrogen peroxide formation |
Resveratrol |
|||
|
Coffea spp. | ↓TC, ↓TG, ↓ALT, ↓AST, ↑Nrf2, ↑HO-1, ↑SOD, ↑GPx, ↓ROS, ↓FAS, ↓SREBP-1c, ↓IL-1, ↓IL-6, ↓TNFα, ↑TNFβ, ↑SIRT1-AMPK pathway, ↓NF-kB |
↑STAT-3, ↓TLR4 [13] |
↑SIRT3, ↑AMPK, ↓PPAR-α, ↓SREBP-1c, ↓ROS, ↑Bcl-2, ↓Bax, |
||
] | [ |
Curcumin |
↑HDL-C, ↓ALT, ↓AST, ↓TG, ↑Leptin, ↑Nrf2/FXR/LXR pathway, ↓CYP3A, ↓CYP7A, ↓CD4+, ↓Leptin, ↓ACC, ↓FAS, ↓SREBP-1c, ↓C/EBP, ↓PPAR, ↓SCD, ↑HO-1, ↓ROS, ↑AMPK/mTOR pathway |
||
|
Red rice (Oryza sativa) | [ |
↓TC, ↓TG, ↓HMG-CoA reductase, ↓ROS, 84] |
|||
[ |
Alkaloid components |
||||
|
Artichoke (Cynara scolymus) |
↓ALT, ↓AST, ↓ROS, ↓MDA, ↓8-deoxyguanosine, ↓CoQ9, ↓GSH |
Berberine |
|||
|
Soy (Glycine max) | ↓TC, ↓TG, ↓ALT, ↓AST, ↑FXR, ↓SREBP-1c, ↓FAS, ↑SOD, ↑GSH, ↓MDA, ↓TNFα, ↓IL-1, ↓IL-6, ↓NF-kB, ↑Nrf2/ARE, ↓TLR4, ↓MyD88, ↑MTTP, ↑CPT-1a, ↑GCK |
↓TG, ↓ALT, ↓AST, ↓ROS, ↑SOD, ↑CAT, ↓FFA, ↑UCP2 |
|||
[ |
Betaine |
↓TC, ↓TG, ↓FFA, ↓ALT, ↓AST, ↑GSH, ↑GPx, ↓NAFLD activity score, ↑SOD, ↑CAT, ↑MDA, ↓ROS, ↓TLR4, ↓NF-kB, ↓IL-1, ↓IL-6, ↓MAPK, ↓FOXO-1 |
|||
|
Alfalfa (Medicago sativa) |
↓LDL, ↑2-deoxy-glucose, ↑glycogen, ↓HMG-CoA reductase, ↓ACAT2 |
[93] |
|||
[ |
Terpenes components |
||||
|
Bergamot (Citrus bergamia | |||||
) |
↓TC, ↓TG, ↓LDL, ↓ROS, ↑HDL, ↓Cardiomyocyte death, ↑autophagy, ↑SOD, ↑GPx, ↓MDA, ↓TNF |
[149]206[150][151][152][153][154][155][156][157][158][159][160][,207161,208],209[162,210][163][,213,214,215,216,217,218,219,220,221,222] |
Celastrol |
↓TG, ↓FFA, ↓ALT, ↓SREBP-1c, ↑AMPKα, ↑LKB1, ↓NF-kB, ↑SIRT1, ↓TNFα, ↓IL-1β, ↓IL-6, ↓ROS, ↑Nrf2/HO-1 |
|
|
Rosemary (Rosmarinus officinalis) |
↓ALT, ↓AST, ↓TAG, ↓FFA ↑AMPK, ↑PPAR, ↓SIRT1/p66shc pathway |
[164][165][166][167][168][169][170][171][172][225,226,227,228,229,230,231,232,233] |
Boswellic acid |
||
|
Peppermint (Mentha piperita) |
↓TC, ↓ALT, ↓AST, ↓TNFα, ↓IL-6, ↓iNOS, ↑UCP1, ↑CPT1, ↓NF-kB, ↓TGFβ, ↓COX-2 |
↓TC, ↓LDL, ↓TAG ↓CYP2B9, ↓Leptin, ↓ROS, ↓lipogenesis, ↑PPAR, ↓SREBP1 |
|||
[ | ||
] | ||
[ | ||
] | ||
[ | ||
] | ||
[ | ||
, | ||
, | 236] |
|
|
Sage (Salvia officinalis) |
↓TC, ↓LDL, ↑HDL, ↓ALT, ↓AST, ↑PPAR, ↓Lipase |
|
|
Hot pepper (Capsicum annuum) |
↑TRPV1, ↑PPAR ↓IL-6, ↓TNF-α, ↓MCP-1, ↓COX-2 |
[180][181][182][[187][241,242183][184][185][,243186,244,245,246],247,248] |
↑: Increase, ↓: decrease of expression, concentrations, or activity. 4-HNE: 4-hydroxynonenal; ACAT2: acetyl-CoA acetyltransferase 2; ACC: acetyl-CoA carboxylase; ACO: acyl-CoA oxidase; AKT: serine/threonine kinase 1; ALT: alanine aminotransferase; AMPK: adenosine monophosphate-activated protein kinase; ARE: antioxidant responsive element; AST: aspartate aminotransferase; CAT: catalase; CASP: caspase; CoQ9: coenzyme Q9; CPT-1a: carnitine palmitoyl transferase-1; CYT: cytochrome; FAS: fatty acid synthase; SCD-1: stearoyl-CoA desaturase 1; FFA: free fatty acids; FOXO-1: forkhead box protein O1; GCK: germinal center kinase; GPx: glutathione peroxidase; GSH: glutathione; HO-1: heme oxygenase-1; IL: interleukin; IRE1a: inositol-requiring enzyme 1a; LDL: low-density lipoprotein; MDA: malondialdehyde; MPO: myeloperoxidase; MTTP: microsomal triglyceride transfer protein; Nrf2: Nuclear factor erythroid 2-related factor 2; PI3K: phosphoinositide-3-kinase; PPARα: peroxisome proliferator-activated receptor α; ROS: reactive oxygen species; SIRT1: silent mating type information regulation 2 homolog 1; SOD: superoxide dismutase; SREBP-1c: sterol regulatory element binding protein-1c; TC: total cholesterol; STAT3: Signal transducer and activator of transcription 3; TAG: tryacylglicerol; TLR4: toll-like receptor 4; TG: triglycerides; TGF: transforming growth factor; TNF: tumor necrosis factor; TRPV1: Transient receptor potential vanilloid 1; TRX-1: thioredoxin-1; UCP1: uncoupling protein-1; VLDL: very low density lipoprotein; WAT: white adipose tissue; and XBP1: X-box-binding protein 1.