Melanocortins, a group of cleavage peptide products of pro-opiomelanocortin (POMC), activate melanocortin receptors on the surface of a diverse range of cell types, leading to different biological actions. They are so named because of their melanotropic activity, that is, the ability of melanocortins to increase pigmentation in melanocytes in the skin and hair follicles, increase concentrations of eumelanin and prevent an increase in photosensitive pheomelanin. Melanocortins are produced by POMC neurons in the pars intermedia of the pituitary gland, the hypothalamic arcuate nucleus and the dorsal medullary nucleus of the solitary tract. They can be distinguished by the presence of an invariant amino acid sequence in each melanocortin peptide, His-Phe-Arg-Trp. The melanocortins produced in humans include alpha-melanocyte stimulating hormone (α-MSH), beta-melanocyte stimulating hormone (β-MSH), gamma-melanocyte stimulating hormone (γ-MSH) and adrenocorticotropic hormone (ACTH).
1. Melanocortins and the Control of Neural Energy Homeostasis
Perhaps the most obvious link between melanocortins and metabolism is their effect on energy homeostasis via hunger reward pathways in the brain (see
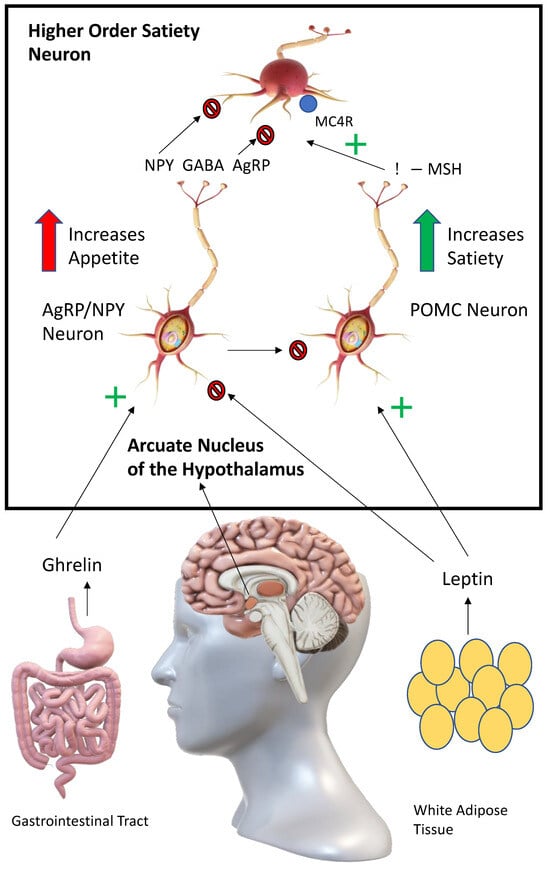
Figure 1). Appetite and food intake is controlled in the arcuate nucleus of the hypothalamus by the competing actions of pro-opiomelanocortin (POMC) and agouti-related peptide/neuropeptide Y (AgRP/NPY) neurons
[1][15]. Activation of POMC neurons leads to anorexigenic effects, while activation of AgRP/NPY neurons leads to orexigenic effects. The way this system functions after a meal is as follows: leptin, which is produced by adipocytes and enterocytes, activates POMC neurons in the arcuate nucleus
[2][16]. This leads to the release of melanocortins (predominantly α-MSH) from POMC neurons which activates melanocortin receptors (predominantly MC4R) on higher order satiety neurons. Satiety and the desire to perform physical activity are increased. In post-prandial periods, the AgRP/NPY neurons act as antipodes of POMC neurons, leading to unpleasant hunger pains. Ghrelin, a hormone produced by the enteroendocrine cells, activates AgRP/NPY neurons in the arcuate nucleus
[3][17]. These neurons, in turn, release AgRP, NPY and GABA (gamma-aminobutyric acid) to antagonise, directly and indirectly, higher-order satiety neurons and their receptors (e.g., MCR4), leading to hyperphagia. AgRP/NPY neurons also directly inhibit the release of melanocortins from POMC neurons during post-prandial periods. Agouti-related protein/peptide (AgRP) is a neuropeptide synthesised by the cell bodies of AgRP/NPY neurons in the ventromedial arcuate nucleus in the brain. Agouti protein is a paracrine competitive antagonist of MC1R and MC4R. The effect of this blockage is hyperphagia, obesity and eventual type II diabetes
[4][18]. Therefore, providing exogeneous melanocortins is likely to correct hyperphagia and restore satiety.
Figure 1. Control of appetite by opposing actions of POMC and AgRP/NPY neurons in the arcuate nucleus. Signalling controlling appetite and food intake in the arcuate nucleus. Following stimulation by ghrelin, AgRP/NPY neurons release AgRP, NPY (Neuropeptide Y) and GABA (Gamma-Aminobutyric acid) to directly and indirectly antagonise downstream satiety neurons and their receptors, including MC4R, increasing feeding. AgRP/NPY neurons also directly inhibit POMC neurons. Following stimulation by leptin, POMC neurons release melanocortins (including α-MSH) to act at MC4 receptors on downstream satiety neurons, increasing satiety.
2. Melanocortins and Leptin: A Potential Downstream Advantage
Since the first ob/ob mouse was discovered by chance in 1949 at the Jackson Laboratory, scientists have been intrigued by the possible use of leptin as an obesity therapy to prevent type II diabetes
[5][19]. Ob/ob have a recessive gene mutation that prevents them from producing leptin
[6][20]. This leads to uncontrollable eating and high blood glucose levels, creating phenotypically corpulent mice. While leptin has been found to have some positive effects in insulin-deficient type I diabetes, it has not seen the same success in treatment for obesity, metabolic syndrome and type II diabetes
[7][21]. This is due to a key difference between ob/ob mice and human type II diabetics. Leptin mutations in humans are vanishingly rare and therefore, most type II diabetics have adequate leptin levels. Unfortunately, in type II diabetics, there seems to be a so-called leptin insensitivity, which is comparable to insulin resistance seen in type II diabetics and patients with metabolic syndrome. As explained in the previous section, melanocortins are downstream from leptin signalling and there is no known resistance to the effects of the melanocortins. Administering melanocortins potentially bypasses the leptin pathway by cozening the appetite centres in the brain into thinking a large calorie-dense meal has just been consumed.
A level of extended satiety can thus be reached, allowing for an amelioration of obesity and its consequent conditions, such as type II diabetes and its associated insulin resistance. The administration of various synthetic melanocortin peptides has been successfully used in this manner in multiple animal and human studies
[8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][6,7,8,9,11,12,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41].
3. An Additional Benefit: The Effects of Melanocortins in the Periphery
The metabolic benefits of melanocortins do not cease at the neuroendocrine level. Melanocortins also directly act on adipocytes via MC4R in the periphery to increase lipolysis and activation of brown adipose tissue (BAT).
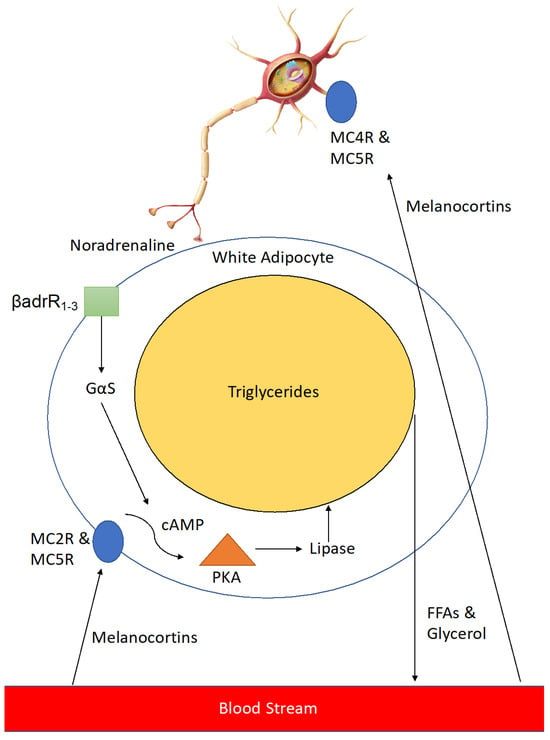
Melanocortins activate neurons innervating adipose tissue via MC4R and MC5R to release noradrenaline which activates β 1–3 adrenergic receptors on the cell surface, leading to a downstream signalling cascade which results in expression of lipase enzyme that breaks down triglycerides in the large lipid droplets found in adipocytes into free-fatty acids and glycerol (
Figure 2). This leads to reduced lipid volume in the adipocytes and thus a reduction in their size, resulting in a loss of adipose tissue mass. In addition, melanocortins help to counteract adipogenesis caused by enhanced levels of Agouti-signalling protein (ASIP) seen in type II diabetics
[34][42].
Figure 2. Stimulation of Lipolysis in white adipose tissue (WAT) by melanocortins. Melanocortins stimulate lipolysis in peripheral white adipose tissue through two related, yet distinct processes. In the first process, following stimulation by melanocortins (including α-MSH) at cell surface G-protein coupled receptors (MC2R & MC5R), an intracellular signalling pathway is mediated through cAMP and protein kinase (PKA), leading to the release of the lipolytic enzyme lipase, which in turn breaks down triglycerides into free fatty acids (FFAs) and glycerol which are then exported into the bloodstream to be metabolised. In the second process neurons innervating WAT are activated by melanocortins through MC4R & MC5R to release noradrenaline. Noradrenaline activates β 1-3-adrenergic receptors (βadrR1-3) leading to a similar intracellular signalling cascade as the first process.
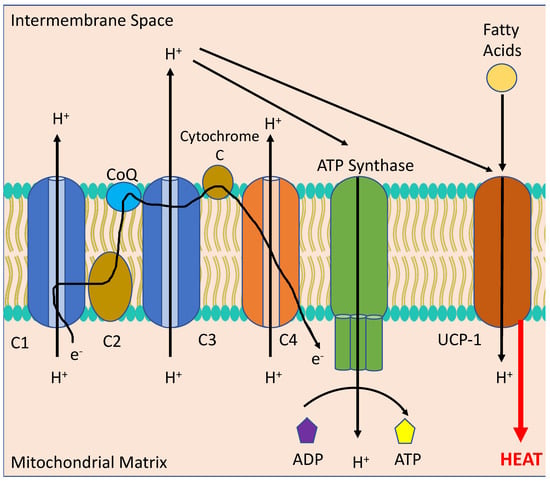
BAT, differentiated from white adipose tissue (WAT), is a metabolically active form of adipocyte found in small quantities in human adults. It is primarily found around cervical, supraclavicular, axillary periaortic, para-vertebral and suprarenal areas. BAT undergoes a process of non-shivering thermogenesis via activation of the mitochondrial protein, uncoupling protein 1 (UCP-1), which acts to decrease the proton gradient across the mitochondrial inner membrane formed by oxidative phosphorylation (see
Figure 3). This scuppers the ability of the mitochondria to produce adenosine triphosphate (ATP) and instead produces copious amounts of heat. BAT is normally activated following a large meal or exposure to cold temperatures. In the early part of the twentieth century an explosive used in munitions factories during World War I, 2,4-dinitrophenol, was repurposed as a weight loss drug due to its mitochondrial uncoupling effects. Unfortunately, whilst being highly efficacious in reducing obesity and insulin resistance, uncoupling drugs tread a very thin line between a therapeutic and a fatal dose
[35][43]. They have been banned by the FDA for use in humans since 1938
[36][44]. Consequently, a direct upregulation or activation of UCP-1 or other uncoupling proteins is considered unwise. Indeed, as this re
svie
arch hw has expounded on, homeostatic mechanisms control the majority of metabolic processes. Therefore, it is crucial for an effective therapy to work within these confines and to only supplement where a rate-limiting step exists. Melanocortins function within these bounds, failing to cause the dangerous cardiovascular side effects of uncoupling drugs while providing a similar metabolic benefit.
Figure 3. Thermogenesis in the mitochondrion of a Brown Adipocyte. Melanocortins stimulate the activation of uncoupling protein 1 (UCP-1) via sympathetic stimulation of neurons innervating Brown Adipose Tissue (BAT) to uncouple the electron transport chain (consisting of C1, C2, Co-enzyme Q, C3, Cytochrome C and C4) from converting adenosine diphosphate (ADP) to adenosine triphosphate (ATP) through ATP Synthase to instead producing heat from fatty acids, leading to reduced body weight.
4. Setmelanotide: Emerging Evidence for the Use of a Clinical Melanocortin Analogue in Metabolic Disease
The practicality of using melanocortins as therapeutics was initially uncertain as bioidentical melanocortin peptides have vanishingly short half-lives and are promiscuous agonists of all melanocortin receptors
[37][45]. Formulation work commenced and biochemists utilised non-canonical amino acid substitutions to prolong half-life and to make the synthetic peptides more specific for certain melanocortin receptors, although some would argue more work needs to be conducted on increasing specificity as every synthetic melanocortin to date is still somewhat promiscuous
[37][45]. Setmelanotide is an example of a designed synthetic selective peptide agonist of MC4R currently being developed by Rhythm Pharmaceuticals as a subcutaneous injectable anti-obesity treatment for Prader–Willi syndrome (PWS) (a genetic cause of MC4R deficiency), leptin receptor deficiency (LEPR), POMC deficiency and lifestyle-induced obesity. Setmelanotide has completed phase III trials for POMC deficiency and LEPR deficiency and was approved by the FDA as first-in-class medication in November 2020. It has also been provisionally approved this year in France for lesional hypothalamic obesity and has been approved in Canada for Bardet-Biedl syndrome. The trials showed significant weight loss (an average of 10%) after one year of setmelanotide therapy
[12][38][39][10,11,46]. The therapy was shown to achieve this 10% average weight loss in 80% of patients with POMC deficiency and 45.5% of patients with a LEPR deficiency
[38][39][40][10,46,47]. Furthermore, it was shown that setmelanotide induced at least a 25% reduction in hunger in 72.7% of patients with LEPR deficiency and in 50% of patients with POMC deficiency
[38][39][10,46]. The average weight loss of patients with POMC and LEPR deficiency was 31.9 kg and 16.7 kg, respectively
[38][39][10,46]. BMI measurements were also significantly lowered by an average of between 22.3 and 49.2% in patients with POMC deficiency and by an average of 10.6% in patients with LEPR deficiency
[38][39][10,46]. Follow up unpublished studies on these patients reported at a recent French paediatrics conference showed sustained weight loss over a period of 3 years. Additional successful phase III trials in other forms of genetic obesity such as Bardet–Biedl and Alström syndromes are currently under priority review with the FDA
[40][41][42][47,48,49]. The FDA approved setmelanotide for Bardet–Biedl syndrome in June 2022. A range of phase II and III trials (titled DAYBREAK and EMANATE) in different rare genetic forms of obesity including MC4R deficiency are currently underway
[9][7]. The mechanism of action of setmelanotide is via suppression of appetite, an increase in satiety and an increase in thermogenesis, leading to a reduction in excess feeding behaviours and a higher resting energy expenditure rate
[9][12][39][40][41][42][7,11,46,47,48,49]. Setmelanotide exerts its effects by binding to MC4R in the PVN of the hypothalamus and the lateral hypothalamic area (LHA). Some have raised concerns of negative neuropsychiatric effects of setmelanotide such as an increased suicidal ideation seen in some patients in follow up studies of patients from the initial phase III study
[42][49]. However, it should be noted that many patients with obesity report low self-esteem, poor body image, depression and anxiety, with depression being a major risk factor for developing obesity
[41][43][48,50]. Therefore, it is unclear whether pre-existing depression is causing the suicidal ideation or if it is worsened by the drug. The manufacturer denies that these cases were a result of treatment, and further follow-up studies report improved quality of life (QOL) assessments in the majority of patients treated
[41][43][48,50]. However, further studies should focus on this potential concerning side effect.
Similar to how GLP-1 analogues are currently used to treat both type II diabetes and obesity (although this re
svie
arch w does not intend to make a direct comparison between the two with regard to either efficacy or side effect profile), melanocortins with similar actions to setmelanotide have potential for treating diabetic patients in the future and for preventing many more patients suffering from obesity, PCOS and metabolic syndrome from developing type II diabetes. Although setmelanotide and other synthetic melanocortins have been developed for genetic forms of obesity, melanocortins (melanotan I and II and bremelanotide) also work to decrease body fat in both healthy non-obese and diet-induced obese individuals
[44][45][46][51,52,53]. A truncated form of MSH caused an average approximate body fat loss of 2 kg over a 6-week period using twice daily nasal administration in healthy, non-obese subjects
[44][51]. These are the only human trials to date using melanocortins in non-genetic forms of obesity. More long-term human studies are needed to determine the safety and utility of melanocortins for treating diet-induced obesity. Various diet-induced obese animal models have shown substantial weight loss after administration of various bioidentical and synthetic melanocortin peptides
[19][22][47][48][27,30,54,55]. This melanocortin-induced weight loss has also been seen even in leptin resistant animals
[49][50][56,57]. With the paradigm-shifting success of GLP-1 agonists, the question may be asked as to what further benefit could melanocortins provide in the obesity treatment milieu. Although they provide a sustained weight loss (up to 15% average of total body weight in long-term large-scale trials), according to studies, 70% of patients taking GLP-1 agonists discontinue use after 24 months (it is still a matter of speculation as to why this is so, but some have postulated that severe nausea and other gastrointestinal side effects could be major contributors, as well as cost and ease-of-access issues)
[51][52][53][58,59,60]. Melanocortins can cause skin darkening, flushing, increased libido, spontaneous erections and mild nausea but are generally well-tolerated, some of these side effects may be seen as additional benefits (although they may not be desirable for all people), hence the colloquial moniker of the melanocortin class as being “Barbie drugs” as they cause skin tanning, weight loss and sexual arousal
[54][55][61,62]. Spurred on by weight loss results seen in healthy obese women using the FDA-approved female sexual dysfunction melanocortin drug bremelanotide (an average loss of 1.3 kg bodyweight over 16 days and a reduction of an average of 400 kcal in food consumption per day in the first phase I trial and an average loss of 1.7 kg bodyweight over 12 days in the second phase I trial; side effects in the trial were classed as mostly mild, whilst a smaller subset of subjects had side effects classed as moderate; skin darkening was reported as a side effect in 15–63.3% of the bremelanotide-treated group across both studies; and five patients had to withdraw due to nausea, dizziness and hypertension), Palatin Pharmaceuticals is developing a combination treatment of bremelanotide and a low-dose GLP-1 agonist to treat obesity
[45][52]. They hope to have improved tolerability and patient compliance with similar or enhanced weight loss compared to GLP-1 agonist monotherapy. Therefore, rather than replacing the already efficacious GLP-1 agonists, in the future, melanocortin/GLP-1 agonist dual therapy may be a way of maximising the potential benefits of both drugs whilst allowing far lower dosages and thus decreasing the chances of side effects and increasing patient adherence.