Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Patricia Guerrero Ochoa and Version 2 by Peter Tang.
Antineoplastic therapies for prostate cancer (PCa) have traditionally centered around the androgen receptor (AR) pathway, which has demonstrated a significant role in oncogenesis. Cholesterol is one of the lipids that has great relevance due to its role in the structure of the cell membrane as well as in signaling pathways linked to the AR pathway, not only due to its precursor role in the generation of androgens, which are the main AR ligands currently used as a target for therapies.
- prostate cancer
- cholesterol
- statins
- mevalonate pathway
- antitumoral
1. The Role of Cholesterol in Plasma Membrane Lipid Rafts
The dynamic functionality of the plasma membrane is underpinned by specialized regions rich in cholesterol and sphingolipids, known as “Lipid Rafts” (LRs), which have the capacity to modulate the accessibility of proteins to regulatory or effector molecules. Cholesterol constitutes approximately 30% of the lipids in the plasma membrane and plays a pivotal role in closely packing the acyl chains of phospholipids, thereby promoting phase separation and stabilizing the formation of LRs [1][42]. Cancer cells exhibit higher levels of intracellular cholesterol and LRs compared to their normal counterparts [2][43]. For instance, in PCa, cell lines like PC-3 have been found to possess LRs at levels five times greater than those in normal cells [3][44].
Lipid rafts play a crucial role in sustaining proliferative signaling by hosting receptors organized within a structure referred to as CASMER, short for “cluster of apoptotic signaling molecule-enriched rafts,” where death receptors are concentrated [4][5][6][45,46,47]. This implies that both types of signals, those promoting cell survival and those leading to cell death, are interconnected in a dynamic manner. CASMER communicate with other subcellular structures to facilitate the transmission of apoptotic signals or induce signals related to cell survival, proliferation, inflammation, cancer growth, and metastasis. In the context of mTOR complex 1 (mTORC1) signaling, molecules like insulin-like growth factor (IGFs) and the epidermal growth factor (EGF) are involved [7][48].
Cholesterol is necessary to maintain lipid raft structure since the removal of cholesterol renders these structures non-functional and leads to the dissociation of proteins from the rafts [6][8][47,49]. Edelfosine, an ether-phospholipid with a high affinity for cholesterol, has emerged as an antitumor drug that acts through lipid rafts, promoting cell death by co-clustering raft-associated Fas/CD95 receptors and inducing apoptosis in a wide range of tumor cells [7][48]. PCa cells induce senescence in a subset of immunosuppressive neutrophils by secreting apolipoprotein E (APOE), which binds to the Triggering receptor expressed in myeloid cells 2 (TREM2). Senescence arrest cell proliferation in a stable manner but cells remain metabolically active, resist apoptosis and induce tolerogenic state in T cells. APOE belongs to a family of lipid-binding proteins that serve as major transporters of systemic cholesterol and triglycerides. The APOE/TREM2 axis correlates with a poor prognosis in PCa patients. Specifically eliminating senescent immunosuppressive neutrophils using histone deacetylase inhibitors enhances the effectiveness of standard therapeutic approaches [9][50].
2. Growth Factor Receptors Reside Inside Lipid Rafts
In the context of PCa, elevated cholesterol levels promote disease progression by inducing the epithelial-to-mesenchymal transition (EMT) through the activation of the extracellular-regulated protein kinases 1/2 (ERK1/2) pathway, mediated by the EGFR, and the accumulation of adipocyte plasma membrane-associated proteins (APMAPs) in LRs. Notably, high cholesterol levels in LRs inhibit EGFR internalization. Consistently, both APMAP mRNA and protein levels are elevated in clinical PCa samples [10][51]. HER2, also called EGFR 2, has been detected on the cell surface of quiescent (G0) PCa cells, which often exhibit resistance to chemotherapy and are associated with recurrence following dormancy [11][52].
Another member of the EGFR family, HER3, has been found to be overexpressed in human CRPC samples, correlating with enhanced proliferation and survival. Notably, these effects can be counteracted by a neutralizing anti-neuregulin 1 (NRG1), which serves as the ligand for HER3 [12][53]. NRG1 is often generated by fibroblasts. In addition to these receptors, G protein-coupled receptors (GPCRs), particularly the G protein-coupled estrogen receptor (GPER), are detected in the cytoplasm of basal epithelial cells, as well as in Gleason patterns 2 or 3, but not in luminal secretory epithelial cells. Various mechanisms associated with GPCRs have been reported in PCa, either alone or in conjunction with estrogen receptors (ERs) [13][14][15][54,55,56].
Similarly, lipid rafts (LRs) are significantly enriched in cancer stem cells (CSCs) compared to non-stem cancer cells, and key CSC markers, such as CD24, CD44, and CD133, are located within these lipid rafts, underscoring their pivotal role in CSCs. LRs play a crucial function in facilitating CSC self-renewal, the EMT, drug resistance, and the CSC niche [16][17][57,58]. In the context of PCa, CSCs exhibit heightened expression levels of CD44 and CD133 [18][59]. In vitro assays have demonstrated that cholesterol depletion disrupts lipid rafts, inhibits cancer cell migration, and abolishes β-catenin activation and the CSC phenotype [17][58], emphasizing the involvement of cholesterol in regulating phenotypic plasticity.
3. Interaction of Cholesterol with Androgen Receptor Signaling, the Critical Route of Prostate Cancer
AR expression has been detected in nearly all primary and metastatic PCa cases, regardless of stage or grade, and is sustained in the majority of androgen-independent PCa and CRPC. Intense nuclear AR staining in CRPC bone metastases is associated with a worse clinical outcome [19][20][60,61]. Some CRPC cases that emerge in patients are linked to the hyperactivation of the androgen signaling pathway due to various factors, including AR gene amplification, AR mutations, the expression of constitutively active AR splice variants (with AR-V7 being the most common variant found in 75% of metastatic PCa cases), intratumoral androgen synthesis, the altered expression and function of AR coactivators, and the signaling crosstalk with other oncogenic pathways [21][22][23][1,62,63]. AR mutations have been identified in 60% of metastatic tumors [24][25][64,65] and 1% of primary PCa cases [25][65]. Functionally, these mutations enable antiandrogens to act as AR agonists and activate AR through estrogens, progesterone, dehydroepiandrosterone (DHEA), androstenediol, and glucocorticoids [26][27][28][66,67,68]. Studies in both patients and animal models have demonstrated that certain point mutations convert AR antagonists into potent agonists [29][69]. AR-V7 is an aberrantly spliced mRNA isoform of AR, leading to a protein lacking the C-terminal ligand-binding domain but retaining the transcriptionally active N-terminal domain. Despite its inability to bind ligands, AR-V7 remains constitutively active in a ligand-independent manner and can drive the growth of CRPC [22][62]. Also, APUC genes have been described as important predictors of clinical response in CRPC, belonging to a family characterized by androgen production, uptake, and conversion, based on genomic analyses of patient germlines [30][70].
Cholesterol derived from lipid rafts in PCa cell lines has been demonstrated to interact with the N-terminal domain of testosterone-activated AR and form complexes with transient receptor potential melastatin 8 (TRPM8). This interaction inhibits the function of TRPM8, a non-selective cation channel involved in the calcium signaling of tumor and stromal cells and has been proposed as a molecular target in cancer development and progression [31][32][33][71,72,73]. This interaction significantly enhances the migratory capacity of PCa cells, promoting invasion and metastasis. Activated AR has a dose-dependent regulatory effect on the function of the TRPM8 channel [34][74], in addition to its known ability to induce the transcription of TRPM8 through binding to androgen response elements (AREs) [35][75]. Likewise, prostate-specific antigen (PSA) has also been observed to modulate TRPM8, facilitating its accumulation in the plasma membrane, similar to the function described for AR but mediated by a G protein-coupled membrane receptor and AKT signaling associated with activated AR [36][37][76,77]. However, various findings suggest that additional factors might modulate the cell response to the TRPM8 channel [32][72]. It is plausible that these factors collectively influence the TRPM8 channel response in PCa [33][38][73,78].
SREBP1 has been implicated in PCa cell proliferation, migration, and invasion. Studies have revealed that an AR/mTOR complex promotes SREBF1 expression and activity, primarily due to the constitutive activation of mTOR resulting from the loss of function of PTEN [39][79]. Moreover, the AR-SREBP1 axis has been identified, forming a self-regulating interaction between the activation of SCAP by AR and SREBP1, which subsequently increases the expression of AR [40][41][80,81]. Androgens are recognized as key activators of SREBPs under both physiological conditions and in steroid-regulated cancers [42][82]. One form of regulation occurs through the stimulation of SCAP expression [43][83], leading to a switch in the isoform expression of INSIG [44][84] and the upregulation of several enzymes involved in fatty acid and cholesterol synthesis under the transcriptional control of AR [45][13]. Perturbations in cholesterol metabolism have been predominantly identified in high-grade tumors [46][3]. Correspondingly, the inhibition of fatty acid synthase (FASN) enhances the immune response to castration-resistant PCa cell line tumors and decreases the expression and transcriptional activity of AR and the AR-variant splicing 7, resulting in a better response to hormone treatment [47][85]. The secretion of cholesterol into the intracellular space is regulated by ATP-binding cassette (ABC) transporters, and data have shown the tissue-dependent regulation of ABC transporters by estrogens, progesterone, and androgens [48][86].
4. The Androgen Receptor–Microbiome–Diet Axis in Prostate Cancer
The gut microbiome is linked with diet; also, the immune response is connected to the modulation of immune function, resulting in a different response to immune checkpoint therapy and serving as a source of testosterone. Men with CRPC have an increased abundance of gut bacteria with androgenic functions. Men with high-risk PCa share a specific gut microbial profile. Most importantly, a high-fat diet causes gut dysbiosis and gut bacterial metabolites such as short-chain fatty acids, propionate, and acetate, which enter systemic circulation, resulting in the promotion of PCa growth; this is called the “gut-prostate axis” [49][50][87,88]. Humans can utilize absorbed acetate and propionate as substrates for lipid, glucose, and cholesterol metabolism [51][89]. Androgen deprivation in mice and humans promotes the expansion of defined commensal microbiota capable of converting androgen precursors into active androgens. The depletion of these microbiota and their replacement with Prevotella stercorea control the growth of PCa [52][90]. Clostridium scindens from fecal human microbiota can convert glucocorticoids into androgens [53][91], and similarly, Ruminococcus gnavus metabolize pregnenolone to androgenic steroids [52][90].
In 2016, the International Agency for Research on Cancer (IARC) recommended the avoidance of weight gain based on a body of evidence from around 1000 studies linking obesity to at least 13 types of cancer. This correlation is often linked to higher levels of lipids in the bloodstream [54][92]. Obesity and altered lipids are associated with the induction of vasculature formation and inflammation through the ETS transcription factor ERG, a gene that encodes key regulators of embryonic development, cell proliferation, angiogenesis, inflammation, and apoptosis. In PCa, the protein ERG contains an ETS DNA-binding domain and a PNT (pointed) domain which is implicated in the self-association of chimeric oncoproteins like TMPSSR2-ERG and NDRG1-ERG [46][55][56][3,93,94]. Individual metabolites correlate with Gleason scores and impact the translocation of ERG, highlighting altered fatty acid oxidation. Specifically, cholesterol has been shown to be negatively correlated with Gleason score [46][3]. In fact, metabolomic differences between Gleason 4 + 3 and Gleason 3 + 4 have been described, suggesting that lipids could be considered markers of PCa aggression [57][95], which is related to the identification of metabolic signatures of PCa [46][58][3,5], similar to other new personalized signatures developed in early PCa based on multimodal biomarkers [59][96].
5. The Role of the cGAS–Sting Pathway in Prostate Cancer
The cGAS–STING pathway plays an important role in the immune defense against both infections and cancer. Upon the activation of the STING protein, various signaling cascades are initiated, ultimately leading to the activation of interferon regulatory factor 3 (IRF3), nuclear factor-kappa B (NF-κB), and the expression of interferon (IFN) and autophagy [60][97]. Acting as a vital component of the genomic stability system within the cytosol and endolysosomal compartments, the cyclic GMP-AMP synthase (cGAS) detects double-stranded DNA (dsDNA) and functions as an adenosine monophosphate (AMP) synthase [61][98].
In the context of PCa, which is often characterized as immunologically “cold” due to limited responses to checkpoint inhibitory therapy, the significance of the STING pathway is underscored. STING activation leads to the upregulation of genes associated with antigen presentation, Th1 chemokine signaling, interferon response, and the expression of programmed cell death protein 1 (PD-L1). These functions of the STING pathway imply its potential relevance in augmenting the immunogenicity of PCa and enhancing the efficacy of immune-based therapies. Consistent with these observations, the enhanced expression of enhancer of zeste homolog 2 (EZH2), a negative regulator of the STING pathway, has been linked to the heightened intratumoral trafficking of activated CD8+ lymphocytes (LT), an increase in M1 tumor-associated macrophages (TAMs), and the reversal of resistance to PD-1 checkpoint therapy [62][99]. These findings are in consonance with Speckle-type POZ protein (SPOP), another modulator of dsDNA, which is mutated in 15% of PCa patients. SPOP is crucial for regulating the balance between immunosuppressive and antitumor activities downstream of STING. It can shift the pathway toward antitumor cGAS–STING–IFN-β signaling [63][100]. Also, in vivo assays suggest that the MVA pathway and IFN signaling pathway are part of a metabolic circuit, sensing each other [64][101].
Recent research efforts have focused on utilizing the cGAS–STING pathway as a therapeutic target for the treatment of PCa, and it has been demonstrated that ionizing radiation can activate this pathway and enhance the response of CD8+ lymphocytes [65][66][102,103]. On the other hand, the cGAS–STING pathway has also been linked to promoting tumor proliferation. Propionibacterium acnes, a common microorganism in normal prostate tissues and PCa, can activate the cGAS–STING pathway and induce the expression of interferons, which subsequently promotes the growth of PCa [61][98].
6. Function of Stroma Elements in Prostate Cancer
Cancer-associated fibroblasts (CAFs) represent the predominant stromal cell type in the tumor microenvironment (TME) [67][68][104,105], and their abundance is notably pronounced in PCa [69][106]. These cells, primarily derived from tissue-resident fibroblasts and other local cells, including tumoral epithelial PCa cells [67][68][70][71][72][73][104,105,107,108,109,110], exert a significant influence on tumorigenesis. Notably, only grafts containing initiated prostate epithelium and CAFs generate tumors [74][75][111,112]. CAFs induce angiogenesis, immunosuppression, metastatic progression, and resistance to therapy. However, in certain contexts, CAFs have been linked to exerting antitumoral activity during the early stages of tumorigenesis [76][113]. This finding aligns with recent research indicating rapid epigenetic remodeling within hours of tumor-specific CD8+ T lymphocytes after sustained exposure to tumor antigens, resulting in a persistent dysfunctional state despite robust activation and proliferation [77][114].
New evidence suggests that tumor cells can circumvent the constraints posed by androgen deprivation therapy (ADT) through the acquisition of glucosamine from CAFs. This process leads to an elevation in O-GlcNAc levels and, ultimately, triggers the induction of 3β-Hydroxysteroid dehydrogenase-1 (3βHSD1), the crucial enzyme responsible for the conversion of dehydroepiandrosterone (DHEA) to the highly potent androgen dihydrotestosterone (DHT) [78][115]. Additionally, mutations occurring in the AR’s ligand-binding domain cause the expanded responsiveness of the AR signaling pathway to nonandrogenic steroid molecules and antiandrogens, along with the expression of constitutively active AR splice variants, alterations in the function of AR coactivators, and intricate crosstalk with other oncogenic pathways [21][79][80][1,116,117]. In this context, cholesterol plays a role in regulating the expression of AR cofactors. Elevated levels of SRC-1, SRC-2, SRC-3, and PCAF gene and protein expression are observed concurrently with heightened AR gene and protein expression. This interplay exerts a notable influence on the initiation and advancement of PCa in a murine model of CRPC [80][117]. In agreement with this, the complexity of the AR is further highlighted by the observation that CRPC is associated with AR overexpression due to clonal selective pressure in the androgen-depleted environment, leading to gene upregulation. Notably, androgen-independent cells require 80% less androgens for growth [81][118], and approximately 30% of these cells exhibit high-level amplifications [82][119]. Furthermore, AR mutations have been identified to increase sensitivity to androgens by four orders of magnitude lower than that required for androgen-dependent PCa cells [83][120]. This hyperactivation of the androgen signaling pathway leads to CRPC.
It has been observed that, in PCa, there is a bidirectional relationship between the signaling pathway of AR and the tumor-associated macrophages (TAMs). AR activation has been described as essential for the ability to induce tumor cell death by TAMs, which is why ADT would be counterproductive to the local innate immune response [84][121]. However, when TAMs become pro-tumor, they can transfer cholesterol to tumor cells in vitro by increasing androgen production and AR activation, suggesting the need to limit cholesterol transfer, the MVA pathway, or TAMs as adjuvant therapy in the context of PCa [85][122].
7. Mitochondria in the Metabolic Deregulation of Prostate Cancer
Metabolic alterations induced by oncogenesis in PCa are sustained through mitochondrial activity and the involvement of dynamin-related protein 1 (DRP1), a mediator of mitochondrial fission. Elevated DRP1 levels in both androgen-sensitive and CRPC cell lines are associated with AR signaling, leading to an enhancement of pyruvate transport into the mitochondria. This, in turn, promotes oxidative phosphorylation (OXPHOS) and lipogenesis, which are hallmark characteristics of PCa [21][1]. Similarly, the upregulation of the pyruvate kinase M2 isoform (PKM2) is critical for controlling lipid homeostasis. The regulation of nuclear respiratory factor 1 (NRF1), responsible for the transcription of the endoplasmic reticulum (ER) transmembrane protein 33 (TMEM33), is a key element of this mechanism. As mentioned earlier, the SREBP proteins are localized in the ER, and TMEM33 recruits the RNF5 enzyme to facilitate SCAP degradation and activate the MVA pathway. These findings are associated with the observation that PKM2 is predominantly expressed in PCa cells as opposed to normal prostate cells. The downregulation of PKM2 is linked to a reduction in tumor growth, while the global knockout of PKM2 accelerates the growth of allografted tumors [86][123]. A part of these considerations is shown in a schematic way in Figure 1.
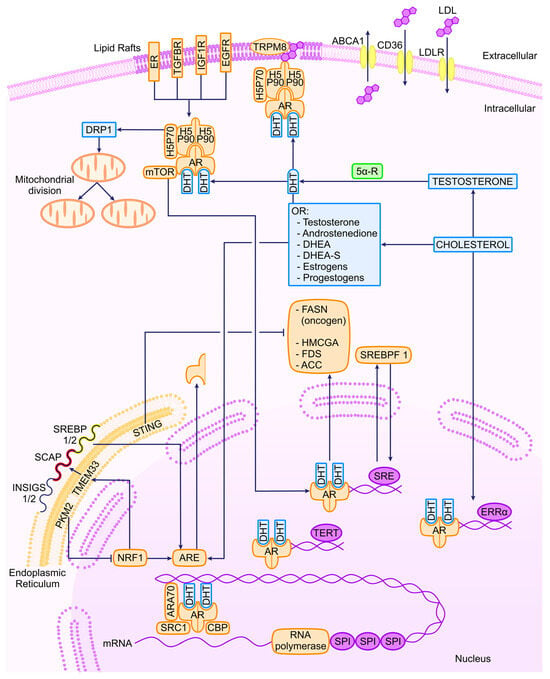
Figure 1. Transcriptional regulators of AR and related molecules in PCa. ER, estrogen receptor; TGFBR, tumor growth factor β receptor; IGF1R, insulin-like growth factor 1 receptor; EGFR, epithelial growth factor receptor; TRPM8, transient receptor potential cation channel subfamily M (melastatin) member 8; ABCA1, ATP-binding cassette subfamily A member 1; LDLR, LDL cholesterol receptor; DRP1, dynamin-related protein; DHT, dihydrotestosterone; 5α-R, 5 alpha receptor; DHEA, dehydroepyandrosterone; PTEN, phosphatase and tensin homolog; cGAS, cyclic GMP-AMP synthase; PKM2, pyruvate kinase M2; TMEM33, transmembrane protein 33; STING, stimulator of interferon response cGAMP interactor 1; NRF1, nuclear respiratory factor 1; ARE, androgen response element.
8. The Telomerase Complex Is Related to Androgen Receptor Signaling
Telomeres are the DNA-protein structures that cap the ends of linear chromosomes and protect them from fusing end to end [87][124]. The telomerase is a protein complex (HSP90, hTERC, diskerin, TEP1, p53, hTERT), the canonical function of which is to protect against telomere shortening [88][125]. The non-canonical functions of the telomerase contribute to cancer progression through apoptosis resistance; an increase in mitochondrial membrane potential and a decrease in ROS; gene expression; and signal transduction [88][125].
In PCa cells, telomere stability may be modulated via an allosteric mechanism involving the AR. The AR interacts with telomeric proteins, specifically TIN2 from the shelterin complex, and exhibits the capability to disrupt these interactions in AR-positive PCa cell lines when treated with the AR antagonist bicalutamide [87][124]. Additionally, there is evidence suggesting a concentration-dependent impact of androgens on human telomerase reverse transcriptase (hTERT) expression. Under physiological androgen levels, characterized by low androgen levels, hTERT expression is induced. Conversely, at high supraphysiological androgen levels, hTERT expression is suppressed by the inhibitor of growth 1 (ING1) and 2 (ING2) [89][126]. Recently, it has been postulated through bioinformatic studies that the AR, zinc finger proteins, and telomeres modulate the global gene expression pattern during PCa progression [90][127]. Conflicting findings about the correlation of telomerase expression and clinicopathological indicators may be consistent with noise caused during processing of tumor tissues by surrounding normal tissue expressing little or no telomerase activity. [91][92][128,129].
The cholesterol-derived hormone estrogen, specifically 17β-estradiol (E2), has the capacity to stimulate the transcription of the human telomerase reverse transcriptase (hTERT) gene in PCa cells. This induction results in an increase in telomerase activity within a short period. Notably, estrogen receptors (ERs) collocate with the hTERT promoter in PCa cells, indicating a direct interaction between estrogen signaling and the regulation of telomerase activity in PCa [93][130]. The significance of these findings lies in the association of estrogens with dihydrotestosterone biosynthesis through the “backdoor pathway.” This pathway, which is not only implicated in the human testis and adrenal glands but also in the classical pathway for androgen synthesis, has been linked to tumor resistance to castration [94][131].