Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Nevin Varghese and Version 2 by Rita Xu.
Hepatitis B virus (HBV) affects around 300 million people worldwide and is a significant risk factor for the development of hepatocellular carcinoma (HCC). Nucleos(t)ide analog therapy has aided in decreasing mortality from HBV.
- hepatitis B virus
- hepatocellular carcinoma
- nucleoside analog
1. Introduction
Hepatitis B virus (HBV) is a global public health problem with an estimated 350 million chronic hepatitis B carriers, causing 820,000 deaths in 2019 alone [1][2][3][1,2,3]. HBV is endemic in many parts of the world, including Southeast Asia, China, and Africa [4]. It belongs to a family of DNA viruses called hepadnaviruses, which is composed of at least ten genotypes, A–J [2][5][6][2,5,6]. The virion is an enveloped nucleocapsid that delivers an incomplete circular DNA genome into the host cell, initiating viral replication [7]. HBV is a dynamic, hepatotropic virus and its infection has a wide spectrum of clinical manifestations [7]. Fifteen–forty percent of HBV-infected patients develop cirrhosis, liver failure, or hepatocellular carcinoma (HCC) [1][8][1,8]. In fact, HBV is the most common hepatocarcinogen, being accountable for 25% of HCC cases in developed countries and 60% in developing countries [9][10][11][12][9,10,11,12].
2. HBV Pathophysiology and Hepatocarcinogenesis
HBV is transmitted by percutaneous inoculation or transmission of infectious bodily fluids. In high-prevalence areas, mother-to-child transmission is the predominant mode of transmission, while unprotected sex and injection drug use are the common modes of transmission in low-prevalence areas [13][14][13,14]. The incubation period of HBV is between 30 and 180 days. During infection, complete and incomplete viral particles are released into the serum of the host, facilitating viral replication [15]. HBV is a non-cytopathic virus, and the liver damage associated with HBV is caused by the host immune response [16][17][16,17]. During acute infection, host immune cells, most prominently CD8 T cells, kill infected cells, inducing hepatic inflammation [4][18][4,18]. Around 70% of patients with acute HBV have subclinical or anicteric hepatitis, while 30% have icteric hepatitis [19]. The clearance of HBV is mediated by the adaptive immune system and HBV utilizes multiple strategies to evade this line of defense [19][20][21][19,20,21]. The hypo-responsiveness of HBV-specific T cells may also contribute to persistent HBV infection [21]. While recovery commonly occurs in immunocompetent individuals, a small proportion of those infected can progress to chronic HBV infection, which is defined as the presence of HBsAg for greater than six months [22]. There are four phases of chronic hepatitis B (CHB) infection: immune tolerance, immune clearance, immune control, and immune escape/reactivation [23]. In the immune tolerance phase, ALT levels are still low, viral DNA levels are high (usually at least 2,000,000 IU/mL), and there is minimal or no inflammation on liver biopsy [4][18][23][24][25][26][4,18,23,24,25,26]. This phase may last for a few years to around 30 years [27][28][27,28]. In the immune clearance phase, hepatitis B e antigen (HBeAg) is positive, there is intermittent or persistent elevation of ALT levels, elevated HBV DNA levels (at least 2000 IU/mL), and some degree of inflammation or fibrosis on liver biopsy [23][28][23,28]. In this phase, HBV-specific CD8 T cells directly attack infected hepatocytes and recruit other immune cells to the liver, which further exacerbate hepatic injury [4][17][18][26][29][4,17,18,26,29]. During immune clearance, patients may present with flares, which—while often being asymptomatic—may be characterized by signs of acute hepatitis. During flares, ALT levels may be elevated to greater than five times the upper limit of normal [23][30][23,30]. The end of the immune clearance phase and beginning of immune control is marked by seroconversion, which is the loss of HBeAg and development of antibodies to hepatitis B e antigen (HBeAb). Intriguingly, the duration of the immune clearance phase has a critical association with the development of complications; those who seroconvert after the age of 40 have a significantly higher risk of cirrhosis, HCC, and CHB compared to those who seroconverted before the age of 30 [23][31][23,31]. In addition, HBeAg positivity is a known risk factor for HCC [26][32][26,32]. The immune control phase is characterized by lower levels of HBV DNA (usually <2000 IU/mL) and ALT, although HBsAg remains [4][18][23][4,18,23]. Some patients, even after seroconversion, may continue to have moderate levels of viral replication with associated abnormal ALT, leading to eventual reactivation of the immune active phase; this phenomenon is known as immune escape [23][28][33][34][23,28,33,34]. Resolution of infection is indicated by disappearance of HBsAg [4][18][4,18]. In CHB, there are crucial changes in immune cell activity and function involving both the innate and adaptive immune systems that lead to hepatic inflammation and hepatocyte killing [17]. Chronic infection may progress to liver fibrosis, cirrhosis, and HCC [17][35][17,35]. HBV-mediated carcinogenesis is a complex process that involves viral DNA integration into the host genome, ultimately leading to viral manipulation of cell-signaling and proliferation. This leads to a cascade of events which converts normal hepatocytes into malignant cells [36][37][38][39][36,37,38,39]. Oxidative stress associated with viral hepatitis changes the cellular environment in such a way that promotes carcinogenesis. Overproduction of free radicals and reactive oxidative species leads to the upregulation of inflammatory pathways that result in hepatocyte release of cytokines and chemokines that recruit neutrophils, monocytes, and lymphocytes [18][37][40][41][18,37,40,41]. As inflammation persists, immune cells, including macrophages and myeloid-derived suppressor cells, become dysfunctional, further amplifying the pro-inflammatory environment. The chronic inflammatory state leads to compensatory hepatocyte proliferation, which leads to accumulation of mutations that promote cell growth and proliferation which predisposes the host to developing HCC (Figure 1) [40][42][40,42].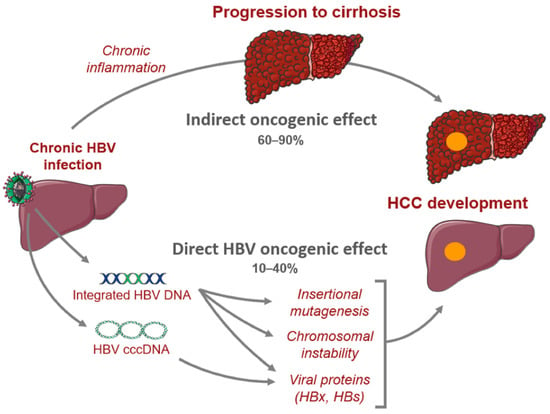
Figure 1. Mechanisms of hepatocarcinogenesis from chronic hepatitis B infection.
3. HCC Risk Factors and Surveillance
HCC is one of the major malignant diseases in the world today and ranks fifth in overall frequency. Its incidence is high in Eastern Asia and sub-Saharan Africa and is increasing in many parts of the Western world [46]. Among cancers, the annual mortality from HCC is relatively high because of its rapid progression and poor prognosis. Unfortunately, the diagnosis is typically made later in its course when therapeutic interventions are generally ineffective. Thus, the focus has been on early screening and treatment of known causes, including chronic hepatitis B which is the most frequent underlying cause of HCC. There are several major risk factors for development of HCC in chronic HBV infection. While having a first-degree relative with HCC, metabolic syndrome, type 2 diabetes, and central obesity are all host factors that have been linked to HCC development in HBV-infected patients or carriers, it appears that liver cirrhosis has been consistently identified to be the most significant risk factor for development of HCC during nucleos(t)ide therapy [47][48][49][50][51][47,48,49,50,51]. Notably, there is increasing evidence that non-alcoholic fatty liver disease (NAFLD) is becoming one of the largest causes of HCC, particularly in industrialized countries [52]. In these patients, HCC can develop even in the absence of cirrhosis. As there are no specific pharmacological therapies for NAFLD, adoption of healthy lifestyle changes including weight loss and regular aerobic physical activity remain the mainstay treatment. Interestingly, bariatric surgery is known to provide durable weight loss and has been identified to be associated with a decreased risk of HCC. A meta-analysis involving nearly 20 million patients showed that bariatric surgery has a protective effect on risk of HCC occurrence and incidence compared to those subjects who did not undergo the procedure [53]. Studies indicate that the presence of male gender, increasing age, higher HBV DNA level, and core promoter mutations are also independently associated with HCC risk [54][55][54,55]. It is postulated that androgens may play a role in the observed difference in incidence of HCC based on gender [56]. Among patients with chronic hepatitis B infection treated with nucleos(t)ide therapy, those with hepatitis D coinfection have been shown to have a nearly sixfold risk of HCC development compared to patients without hepatitis D coinfection [57]. While less investigated, concurrent HCV or HIV infection has also been identified to have an association with increased incidence of HCC [58][59][58,59]. The risk factors for hepatocarcinogenesis can be seen in Figure 2.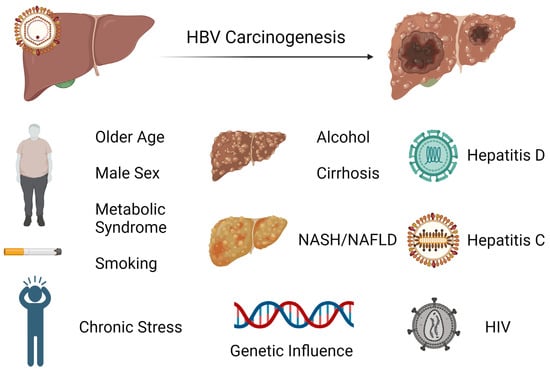
Figure 2. Exogenous and endogenous risk factors for hepatocarcinogenesis from chronic hepatitis B infection.