1. Introduction
The cerebellum has been well established as a subcortical center for motor coordination and motor learning, while its nonmotor functions, e.g., reward and cognition, have attracted increasing attention
[1]. The multifaceted role of the cerebellum relies on its well-organized structure and circuitry during development
[2]. Neurons in the cerebellum are derived from two germinal zones: the cerebellar ventricular zone (VZ) and the cerebellar rhombic lip (CRL)
[3]. The progenitors in the cerebellar VZ give rise to GABAergic inhibitory neurons, including Purkinje cells (PCs), Golgi cells, basket cells, stellate cells, candelabrum cells, and small neurons in the cerebellar nuclei (CN) in mammals
[2]. Likewise, the neuroepithelial cells from the CRL produce glutamatergic excitatory neurons, including granule cells (GrCs), unipolar brush cells, and large projection neurons in the CN
[2]. These GABAergic and glutamatergic neurons are arranged into a three-layer folded cortex (the molecular, PC, and GrC layer, from superficial to deep) with paired sets of CN (fastigial, interposed, and dentate nucleus, from medial to lateral) at its base. During the neuronal morphogenesis and positioning in the cerebellum, PC maturation and GrC proliferation and migration have been recognized as critical events
[4]. PCs migrate from the VZ toward the outer surface of the cerebellum, form a single lamina, i.e., the PC layer, and develop large and complex dendritic trees at the early postnatal stage
[5]. In parallel, CRL gives rise to cerebellar GrC progenitors, which proliferate and migrate to form the external granular layer (EGL), completely covering the embryonic cerebellar surface. GrC progenitors proliferate in the outer EGL to generate the postmitotic GrCs that invade the inner EGL and migrate radially through the molecular layer, past the PC layer, to eventually settle in the internal granular layer (IGL)
[6]. By the end of the third postnatal week, EGL cells have completed migration to the IGL, the GrC layer in the mature cerebellum
[7].
The maturation of the cerebellum leads to the final setting of the cerebellar circuitry. The cerebellar neurons receive two major types of afferent inputs—mossy fibers (MFs) and climbing fibers (CFs)—both of which use glutamate as their primary neurotransmitters
[8]. The MFs, originating from many sources in the brainstem and spinal cord, synapse with GrC dendrites in the GrC layer
[8]. The GrCs then send their axons, i.e., the parallel fibers (PFs), up to the molecular layer, and relay MF inputs to the distal dendrites of PCs
[8]. Each PC receives functionally weak but numerous (e.g., 100,000 in mice) PF synapses
[8]. In contrast, the CFs arise exclusively from neurons in the inferior olive (IO), and innervate the proximal dendrites of PCs
[8]. In the mature cerebellum, each PC is traditionally assumed to be mono-innervated by a single strong CF because of the axonal competition during development
[9]. In addition, multibranched PCs have recently been found to receive more than one CF afferent input
[10]. In either case, PCs integrate information from excitatory inputs of PFs and CFs and local inhibitory input from molecular layer interneurons, e.g., basket cells and stellate cells, and generate the sole output of the cerebellar cortex
[8]. Finally, CN neurons receive inhibitory inputs from PCs as well as direct inputs through MF and CF collaterals, constituting the cerebellar final outputs
[8]. Through this input–output organization, the cerebellum serves as a unique hub in the central nervous system to not only fine-tune motor activity but also modulate nonmotor processes. Moreover, multiple forms of plasticity have been revealed at various synaptic and extrasynaptic sites in the cerebellar cortex and nuclei, which have been considered to be neural substrates for motor learning
[11].
In addition to the classic small-molecule neurotransmitters, e.g., glutamate and GABA, neuropeptides have also been reported to act as critical mediators of intra- and extra-cerebellar circuit connectivity. Ito has summarized 22 different types of neuropeptides distributed in the cerebellum
[12]. A more recent study has determined the expression of neuropeptides in the cerebellum from birth to adulthood by using a semiquantitative peptidomic approach, and identified a total of 33 neuropeptides
[13]. In contrast to classic neurotransmitters, the synthesis, release, and removal of neuropeptides exhibit unique features. Firstly, neuropeptides are synthesized in the somata of neurons and transported down the axon, whereas small-molecule neurotransmitters are synthesized and stored in the terminal for fast release
[14]. Secondly, a longer train of action potentials and more calcium influx are required for neuropeptides to be released into the synaptic cleft, while a single action potential may give rise to the release of amino acids
[14]. In addition, neuropeptides are generally degraded by peptidases located on cytomembranes of neurons and glia. Notably, the concentration of these peptidases is relatively low, so neuropeptides may diffuse from their release site to remote receptors at a long range, allowing them to affect larger populations of neurons
[14]. Unlike neuropeptides, the synaptic effects of small-molecule neurotransmitters are terminated by quick transport back into nerve terminals
[14]. In the cerebellum, multiple neuropeptides have been reported to be essential for cerebellar development and modulation of neuronal activity and synaptic plasticity. Moreover, neuropeptides have also been implicated in the pathological changes in the cerebellum and may serve as potential therapeutic agents in cerebellum-related disorders. Major cerebellar neuropeptides that have attracted the most attention in the last two decades are summarized in
Figure 1 and
Table 1.
Table 1.
Major cerebellar neuropeptides that have attracted the most attention in the last two decades.
2. BDNF
BDNF, discovered in 1982, is a member of the neurotrophic family of growth factors
[15]. Synthesis and maturation of BDNF is a multistage process that culminates in the formation of a mature 119-amino acid protein neuropeptide
[16]. BDNF is distributed in various brain regions, but the cerebellum and hippocampus have the highest expression levels
[17]. In the cerebellum,
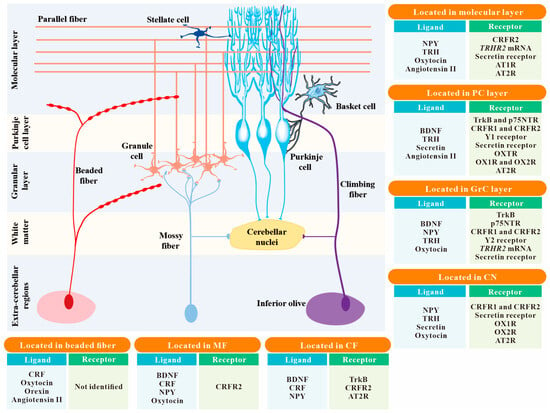
Bdnf mRNA is restricted to the IGL and PCs, and its protein is widely distributed within the cerebellar cortex
[18] (
Figure 1). Moreover, cerebellar BDNF comes from extracerebellar sources via CFs and MFs
[18][19][20][18,19,20]. Two types of receptors binding to BDNF, i.e., tropomyosin-related kinase B (TrkB) and the low-affinity p75 neurotrophin receptor (p75NTR), have been reported to be expressed abundantly in GrCs with a weaker expression in PCs of the cerebellum
[19][20][19,20]. TrkB receptor is expressed in both pre- and post-migratory GrCs
[19], whereas p75NTR receptor is solely expressed in pre-migratory GrC precursors in the EGL and downregulated before GrC precursors migration and differentiation
[21].
Figure 1. The spatial distribution of 8 common neuropeptides and their receptors within the cerebellar circuitry. The localizations of neuropeptides and their receptors in the 7 main structures of the cerebellum, including the molecular layer, PC layer, GrC layer, CN, MF, CF, and beaded fiber, are summarized in the insert boxes. Neuropeptides not mentioned in the text are not listed in these boxes. Abbreviation: PC, Purkinje cell; GrC, granule cell; CN, cerebellar nuclei; MF, mossy fiber; CF, climbing fiber; BDNF, brain-derived neurotrophic factor; CRF, corticotropin-releasing factor; NPY, neuropeptide Y; TRH, thyrotropin-releasing hormone; TrkB, tropomyosin-related kinase B (one of the BDNF receptors); p75NTR, p75 neurotrophin receptor (a low-affinity receptor for BDNF); CRFR1 and CRFR2, CRF receptor 1 and CRF receptor 2; Y1 receptor and Y2 receptor, neuropeptide Y receptor 1 and neuropeptide Y receptor 2; TRHR1 and TRHR2, TRH receptor 1 and TRH receptor 2; OXTR, oxytocin receptor; OX1R and OX2R, orexin type 1 receptor and orexin type 2 receptor; AT1R and AT2R, angiotensin II type 1 receptor and angiotensin II type 2 receptor.
BDNF is essential for cerebellar development. Firstly, many lines of evidence have indicated that BDNF promotes the survival and maturation of GrCs. In cerebellar GrC cultures, GrCs have more survival and fiber outgrowth in the presence of BDNF
[22]. Mice with gene deletion of BDNF exhibit increased apoptosis of GrCs and a sparser and thinner IGL on postnatal day 8 (P8)
[23]. In addition, deletion of BDNF delays the disappearance of EGL
[23], indicating an impaired migration and maturation of GrCs. A recent study has also shown a critical role of p75NTR in the EGL of the cerebellum. In the cultured EGL cells from p75NTR knockout rats, the cell cycle of GrC progenitors is accelerated and thus leads to excessive progenitor proliferation within the EGL
[21]. In contrast to p75NTR, TrkB receptor is abundantly distributed in mature GrCs
[24], indicating that TrkB signaling may promote the maturation and maintenance of differentiated GrCs. In cultured cerebellar EGL cells whose differentiation has been initiated by signals within the cellular reaggregates of EGL, BDNF promotes neurite extension and survival of differentiated GrCs, which can be blocked by the Trk inhibitor K-252a
[24]. However, gene depletion of TrkB receptor in cerebellar precursors does not reduce the number of GrCs or their excitatory synapses when EGL progenitor cells have completed their migration to the IGL by the end of the third postnatal week
[25], indicating that there may be a compensatory mechanism in vivo to lead to a normal final density of GrCs in the adult cerebellum in the absence of TrkB receptor. In line with this speculation, it has been found that TrkB and the neurotrophin 3 receptor TrkC cooperate in promoting the survival of GrCs. Massive cell apoptosis of GrCs has been observed on P14 in TrkB and TrkC double-mutant mice, but not in mutant mice deficient of a single Trk receptor
[26]. Secondly, BDNF signaling is involved in CF synapse elimination in the cerebellum. In BDNF knockout mice, innervation of PCs by multiple CFs fails to be fined into mono-innervation
[27]. By PC-specific deletion of BDNF combined with the knockdown of BDNF receptor in CFs, it has been observed that BDNF acts retrogradely on TrkB in CFs, and facilitates the elimination of CF synapses from PC soma after P16
[27]. Finally, cerebellar BDNF signaling is essential for the development of inhibitory synapses in the cerebellum. In PC-specific BDNF knockout mice, the density of parvalbumin-positive GABAergic interneurons in the molecular layer and the density of GABAergic terminals on the PC soma are markedly reduced on P15 and P16
[27]. By cerebellum-specific deletion of TrkB, the density of GABAergic synapses decreases in both the GrC layer and molecular layer during P50–P80
[25].
Moreover, accumulating studies have highlighted a direct and critical role of BDNF in regulating cerebellar neuronal excitability, synaptic transmission, and synaptic plasticity. In the cerebellum, BDNF has been found to elicit rapid depolarizing responses in GrCs and PCs by activating Na
+ channels coupled to TrkB receptor
[28]. Several independent studies have shown that BDNF exerts dual actions to modulate GABAergic transmission. By activating potassium–chloride cotransporter 2 (KCC2) coupled to TrkB signaling, BDNF reduces the efficacy of GABAergic transmission in PCs in rat cerebellar slices
[29]. Another study has shown that GABAergic transmission from PCs is potentiated by co-activating TrkB and Src (a member of nonreceptor tyrosine kinases)
[30]. In addition, BDNF may also modulate GABAergic transmission by regulating the amount of GABA
A receptors, as well as the synthesis and transport of GABA. The hyperammonemia-induced overexpression of GABA
A receptors, GABA-synthesizing enzymes, and GABA transporters within the cerebellum can be reversed by the blockage of the BDNF-TrkB pathway
[31]. Notably, BDNF also plays a fundamental role in the modulation of cerebellar synaptic plasticity. In cerebellar PCs, BDNF can induce ionic plasticity, a form of synaptic plasticity unique to inhibitory neurotransmission, which is manifested as a decrease in synaptic strength of GABAergic transmission due to a reduced transmembrane CI
− gradient mediated by the coupling between GABA
A receptor and KCC2
[32]. Furthermore, BDNF has been reported to be required for vesicle docking and to contribute to short-term plasticity, such as paired-pulse facilitation, at the PF–PC synapses of the cerebellum
[33].
The suppression of BDNF expression is a potentially momentous phenomenon in many neurodegenerative disorders caused by abnormal expansion of tri-nucleotide (CAG) repeats encoding polyglutamine (polyQ), such as spinocerebellar ataxias (SCAs). The cerebellum of individuals with SCA type 6 (SCA6) shows reduced
Bdnf mRNA expression and abnormal localization of BDNF protein
[34]. Moreover, mice with a targeted gene deletion of BDNF show decreased TrkB signaling in GrCs and PCs and exhibit a wide-based ataxic gait
[23], indicating that the impairment of BDNF-TrkB signaling pathway may not only result from polyQ expansion but also contribute to the onset of ataxia symptoms. Several lines of evidence have suggested that an enhancement of the cerebellar BDNF pathway may provide therapeutic strategies for the treatment of spinocerebellar ataxias. It has been reported that chronic treatment with aminopyridines, which are associated with increased levels of cerebellar BDNF, can correct early dysfunction and delay neurodegeneration in SCA1 mice
[35]. Moreover, an increase in the production of BDNF may act as a primary mechanism by which mesenchymal stem cell therapy promotes PC survival and improves ataxic motor dysfunction in Lurcher mutant mice
[36]. In addition to cerebellar ataxias, BDNF plays crucial neuroprotective and immunomodulatory roles in other pathological situations. In rat cerebellar GrC cultures, BDNF administration markedly reduces the glutamate-induced cell death of GrCs
[22]. Rat pups with subcutaneous administration of estradiol on P4 increase cerebellar
Bdnf mRNA and protein levels, and attenuate ethanol-induced PC loss and motoric impairment, indicating the involvement of BDNF pathway in neuroprotective effects of estrogen against ethanol toxicity in the developing cerebellum
[37]. Enhanced cerebellar BDNF expression has also been reported to contribute to improving high-fat diet-induced dysregulations in cytokines in the cerebellum with exercise training and melatonin administration
[38]. In addition, BDNF underlies the beneficial effects of environmental enrichment
[39], which has been known to be highly related to the reduction in proinflammatory cytokines and chemokines in the brain
[40]. Therefore, the cerebellar BDNF signaling pathway may provide potential therapeutic targets for cerebellar-related neuroimmune and neurodegenerative disorders.
3. CRF
CRF, chemically identified in 1981 and also known as corticotropin-releasing hormone (CRH), is a 41-amino acid polypeptide that is synthesized and released by neurons in the paraventricular nucleus of the hypothalamus, as well as those in broad extrahypothalamic brain regions, including IO, the sole origin of cerebellar CFs
[41][42][41,42] (
Figure 1). CRF is well known to be implicated in autonomic and behavioral components of the stress response
[42]. It has been reported that
Crf mRNA in the IO is upregulated shortly in stress-induced motor responses, and IO-specific knockdown of CRF is sufficient to induce motor deficiency under either normal
[43] or challenging conditions
[44]. In addition to CFs, CRF may also be expressed in MFs and present as a beaded plexus lying parallel to the pial surface, above and subadjacent to the PC layer
[45]. Among these populations of CRF-immunopositive afferent inputs, IO-derived CFs are the biggest source. CRF immunoreactive somata are distributed throughout all divisions of the IO
[45]. Postmortem human cerebellum obtained from patients with olivopontocerebellar atrophy showed a significant decrease in cerebellar CRF concentration
[46], indicating a role of CRF signaling in cerebellar ataxias. In fact, the CF-derived CRF promotes cerebellar motor coordination. By using retrograde tracing, immunohistochemistry, and whole-cell patch clamp recordings, the direct CRFergic projections from the IO have been identified to excite glutamatergic projection neurons in the cerebellar interposed nucleus via two CRF receptors, CRFR1 and CRFR2, coupled to inward rectifier K
+ channels and hyperpolarization-activated cyclic nucleotide-gated channels
[43]. In addition, microinjection of CRF into the bilateral interposed nuclei not only promotes motor performances in normal rats but also ameliorates ataxia-like motor abnormalities induced by the IO-specific
Crf mRNA downregulation and 3-acetylpyridine (3-AP) administration
[43]. These results strongly suggest that CRF in the olivocerebellar system may hold a key position in the pathophysiology and treatment of cerebellar ataxia.
In addition to the excitatory effects on neurons in the CN, CRF signaling is essential for neuronal activity and plasticity modulation of PCs in the cerebellar cortex. CRF increases PC excitability by modulating sodium, potassium, and hyperpolarization-activated action currents
[47]. Antagonizing CRF receptors by α-helical CRF or astressin blocks long-term depression (LTD) of PF–PC
[48] and CF–PC
[49] synaptic transmissions. In contrast to improving LTD, which favors the suppression of glutamatergic transmission in PCs, CRF has also been reported to promote CF–PC glutamatergic transmission, manifested by an enhancement in complex spike activity of PCs, via the presynaptic autoreceptor CRFR2 and the underlying PKA signaling in CF terminals
[50]. These results indicate that CRF may regulate the strength of excitatory inputs to PCs bidirectionally. Moreover, CRF can block facial stimulation-induced LTD at inhibitory synapses between the molecular layer interneurons and PCs by triggering long-term potentiation (LTP) at these synapses via CRFR1 and its underlying PKC signaling pathway
[51]. In addition to the modulatory effects on synaptic inputs to PCs, GrC-specific knockout of CRFR1 has also been shown to affect MF–GrC transmissions, manifested by converting the high-frequency MF stimulation-induced LTP into LTD
[52]. Consistent with its significant roles in plasticity modulation, cerebellar CRF signaling is actively involved in motor learning and adaptation. Mice with GrC-specific CRFR1 deletion show accelerated eyeblink conditioning learning
[52]. Intriguingly, in line with the result that stress disrupts eyeblink conditioning in humans
[53][54][53,54], depletion of CRFR1 in rodents rescues animals from the hazardous effects of early life stress or prolonged stress on learning
[55][56][55,56]. Furthermore, the transcription and expression of CRF within the IO are influenced by optokinetically evoked IO discharge and may contribute to optokinetic adaptation
[57].
CRF and its receptors are present during the critical period of cerebellar development and sustainably expressed throughout the postnatal period
[58][59][58,59]. The cellular localization of CRFR1 immunoactivity in PCs translocates from the apical processes at birth (P0), to the somata (P3), and finally to the primary dendrites (P9) during PC development in the postnatal mouse cerebellum
[60]. In contrast to CRFR1, CRFR2 immunoreactivity is predominantly localized to the somata and axon hillocks of cerebellar PCs, and is not expressed in large quantities until P12
[61]. A truncated isoform of CRFR2 is primarily expressed in presynaptic localization within the cerebellar cortex
[62]. It has been proposed to be involved in the development of extracerebellar afferents
[63]. Several lines of in vitro evidence indicate the role of CRF signaling in the development of PCs. Applying CRF to rat cerebellar slice cultures increases the density of dendritic spines on PCs, which can be blocked entirely by combined administration of selective antagonists for CRFR1 and CRFR2
[64]. Moreover, more dendritic outgrowth and elongation are observed in PCs exposed to CRF intermittently
[65]. Instead, exposure to CRF constantly inhibits the dendritic outgrowth of PCs
[65].
4. Angiotensin II
Angiotensin II is a bioactive octapeptide of the renin–angiotensin system (RAS) involved in controlling blood pressure and maintaining fluid homeostasis that was first isolated in 1954
[66]. This peptide can also be produced within the central nervous system and serve as a neurotransmitter and neurohormone
[67]. In an early study, Changaris et al.
[68] found that fibers with angiotensin II immunoprecipitation traverse through the white matter and diverge within the GrC layer, terminating on the somata of PCs in the rat cerebellum (
Figure 1). Detecting angiotensin II distribution in the rat cerebellar cortex by immunogold staining
[69] has revealed that angiotensin II immunoreactivity is prominent in cerebellar PCs, GrCs, basket cells, and stellate cells. At the subcellular level, the peptide is clearly localized in the transcriptionally active euchromatin of nuclei, suggesting a role of angiotensin II in regulating gene transcription
[69]. By comparing their global transcriptional data with additional published datasets, Elkahloun and Saavedra have reported that the selective blocker for angiotensin II type 1 receptor (AT1R), candesartan, can protect from the alterations in aging- and senescence-related gene expression in the cerebellum of old mice
[70]. As expected, angiotensin II receptors were also found in the cerebellum using an in vivo cryotechnique, albeit with distinct expression patterns. Immunoreactivity of AT1R is highest in the outer molecular layer and largely overlaps with Bergmann glia in the mouse cerebellum
[71] (
Figure 1). Unlike AT1R, AT2R immunoreactivity in the rat cerebellum is primarily associated with the PC layer and the CNs, rather than the molecular layer
[72]. In addition, the levels of two angiotensin II receptors are both time-dependently modulated under hypoxia in the cerebellum
[71], which might be related to the role of angiotensin II and its receptors in regulating cerebellar hemoperfusion.
During the critical period in the postnatal development of the cerebellum,
At2r mRNA is initially discretely localized in neurons of the IO and translated into AT2R in situ, and then transported through CFs to the PC layer of the cerebellar cortex
[73]. The result suggests a role of AT2R in the development of the IO–cerebellar pathway
[73]. In microexplant cultures of the cerebellum from three-day-old rats, specific activation of the AT2R induces major morphological changes, including neurite outgrowth and cell migration, two important processes in the organization of the various layers of the developing cerebellum
[74].
Angiotensin II has also been reported to be involved in regulating neuronal excitability and synaptic transmission within the cerebellum. Microiontophoretic application of angiotensin II specifically suppresses the spontaneous firing of PCs and enhances the inhibition of GABA, which can be antagonized by a specific GABA antagonist, bicuculline methochloride
[75]. In addition, previous studies have suggested that angiotensin II and its receptors are emerging as critical players in neuroinflammation in the cerebellum. In cerebellar astroglial cultures, angiotensin II administration induces a robust inhibitory effect on
Il-10 mRNA expression
[76]. Moreover, angiotensin II-AT1R signaling induces cyclooxygenase 2 expression to stimulate proinflammatory and mitogenic actions in cerebellar astrocytes in vivo, which potentially contributes to central sympathetic overactivity and elevates blood pressure
[77]. In contrast to AT1R, several findings suggest a role for AT2R in development and cognitive function
[78]. Genetic variants in AT2R abundant in the cerebellum have been identified in human X-linked mental retardation
[78], with clinical symptoms associated with autistic spectrum disorder (ASD)
[79]. Notably, central angiotensin II is involved in the positive regulation of systemic oxytocin
[80], which could influence the connections between the cerebellum and forebrain structures associated with ASD
[81].