Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Yuefeng Chu and Version 2 by Lindsay Dong.
Transcription factors are pivotal regulators in the cellular life process. Activating transcription factor 3 (ATF3), a member of the ATF/CREB (cAMP response element-binding protein) family, plays a crucial role as cells respond to various stresses and damage. As a transcription factor, ATF3 significantly influences signal transduction regulation, orchestrating a variety of signaling pathways, including apoptosis, ferroptosis, and cellular differentiation. In addition, ATF3 serves as an essential link between inflammation, oxidative stress, and immune responses.
- ATF3
- inflammatory
- apoptosis
- ferroptosis
- microorganism
1. Introduction
Transcription factors (TFs) play a crucial role in the regulation of cellular function and the development of diseases due to their direct influence on gene expression, thus becoming a focal point of research in recent years. TFs regulate a variety of cellular physiological activities and immune responses by initiating or inhibiting the transcription of specific genes. Serving as key regulatory factors in the coordination of immune responses, TFs are essential for the activation of immune cells and their direct involvement in the production of inflammatory cytokines [1][2][3][1,2,3]. However, in the prolonged struggle between pathogens and hosts, some pathogenic microorganisms have evolved strategies to manipulate host TFs, thereby promoting their survival and replication within the host. Studies have elucidated that specific pathogenic organisms manipulate the activation of the NF-κB pathway as a survival strategy, which can effectively protect the host cell from apoptosis and allow bacterial survival within host cells [2]. Therefore, TFs are not only central to the host immune response but are also a vital strategy for some pathogens to establish infection. Increasing evidence suggests that TFs, such as STAT1, STAT2, JunB, CHOP, ATF3, and NF-κB, are involved in the interaction between host and pathogen and the regulation of host immune responses [2][4][5][6][7][2,4,5,6,7]. Thus, understanding the specific roles of TF in different infectious environments can provide insight for the development of targeted therapies to enhance host defense capability.
Activating transcription factor 3 (ATF3) is a stress-induced transcription factor that belongs to the activating transcription factor/cAMP response element-binding protein (ATF/CREB) family [8][9][8,9], whose members include ATF1, CREB, CREM, ATF2, ATF3, ATF4, ATF5, ATF6, ATF7, and B-ATF [10]. ATF3 regulates gene transcription by forming homodimers or heterodimers through its basic-region leucine zipper (bZIP) domain, thus modulating the biological functions of genes. ATF3 expression is relatively stable under normal physiological conditions, but changes in its expression are associated with various pathophysiological responses such as inflammation, oxidative stress, stress of the endoplasmic reticulum, and cell death [8][9][11][8,9,11]. ATF3 expression is induced by a variety of signals, including those initiated by cytokines, genotoxic agents, or physiological stressors [10]. ATF3 is upregulated under multiple stress conditions, regulating the interaction between cellular metabolism and immune and inflammatory responses, thus maintaining cellular homeostasis.
23. Mechanisms of ATF3 Induction under Stressful Physiological Conditions
ATF3 shares the same binding site, 5′-TGACGTCA-3′, found in other transcription factors of the ATF/CREB family [10]. They interact with target DNA by binding to the entire region within the bZIP domain [8][10][8,10]. Several genes have been identified to possess this recognition sequence, including Nrf2, JunD, c-Jun, and IL-6 [12][13][18,19]. Intriguingly, some promoters of target genes regulated by ATF3 have binding sequences that differ from these common motifs (Table S1). ATF3 expression is relatively stable under normal physiological conditions; however, it rapidly changes in response to disturbances in the internal environment or external stimuli [14][20]. Changes in ATF3 expression are induced by inflammatory responses, cell death, cytokines, and cellular stresses (oxidative stress, DNA damage, or endoplasmic reticulum stress (ERS)). The specific induction mechanism may vary depending on the type of stress but usually involves the activation of stress-responsive kinases and upstream transcription factors, which then bind to the ATF3 promoter and stimulate its transcription [15][16][21,22]. Due to its induction in response to various stress signals, ATF3 is considered a stress-induced gene [17][18][19][23,24,25], which participates in cellular growth, tissue remodeling, cytoskeletal reorganization, and inflammation [15][21]. NF-E2-related factor 2 (Nrf2) transcriptionally upregulates ATF3 expression in astrocytes, thus promoting antioxidant and cytoprotective functions [20][26]. Naringin (Nar) activates ATF3 and inhibits PINK1 transcription by suppressing ERS and mitochondrial autophagy-related genes, as well as the expression of downstream ERS proteins [21][27]. ATF3 interacts with the cAMP response element (CRE) sequences through its basic region and forms homodimers or heterodimers with other CREB family members through its bZip domain [15][22][21,34]. Heterodimers, such as those formed by ATF3 with C-jun, ATF2, and JunB, have been demonstrated to possess transcriptional activation capabilities, enhancing the transcriptional activity of downstream target genes [23][24][25][32,35,36]. Currently, it is widely accepted that ATF3 might occur through stabilizing inhibitory cofactors on the promoter, thereby suppressing the transcriptional activity of downstream genes [13][22][19,34]. ATF3 recruits histone deacetylase 1 (HDAC1) to promoters containing ATF3 binding sites. Subsequently, HDAC1 causes histone deacetylation, leading to chromatin condensation and transcriptional repression [13][26][19,37]. Histone acetylation is a crucial physiological process that opens the chromatin structure, allowing transcription factors to bind to gene promoters and activate transcription [13][27][19,38].34. Implications of PAMPs and PRRs Activation on the Expression of ATF3
The Toll-like receptor (TLR) family consists of pattern recognition receptors (PRRs) involved in the detection of pathogen-associated molecular patterns (PAMPs) [10][28][10,42]. TLR1, 2, 4, 5, and 6 are expressed on the cell surface membrane and recognize bacterial and fungal products, while TLR3, 7, 8, and 9 are located in intracellular endosomes, specializing in the detection of pathogen-associated nucleic acids [10][28][10,42]. Upon recognition of their ligands, TLRs initiate complex cell signaling pathways, conferring antiviral and antibacterial states to the cell and promoting the expression of inflammatory cytokines, chemokines, and co-stimulatory molecules, which are crucial for the activation of adaptive immune responses [10][28][10,42]. Studies have shown that murine bone marrow macrophages treated with multiple PAMPs recognized by TLRs such as LPS, pIC, CpG-ODN, pIC/CpG-ODN, and zymosan significantly increase ATF3 protein expression [29][43], indicating that ATF3 expression is induced by various TLR responses.45. Modulatory Role of ATF3 in Inflammatory Cytokine
As a pivotal hub in the network of cellular adaptive responses and a transcription factor regulating immune response genes, ATF3 is a key regulator of both local and systemic inflammation, aiding cells in responding to disruptions in internal homeostasis. This is due to its ability to positively or negatively modulate the functional status or bioactivities of immune and nonimmune cells. The role of ATF3 has been studied in different contexts. In macrophages, ATF3 is a crucial regulator of innate immune responses. It is a product of TLR signaling and modulates inflammation in lipopolysaccharide (LPS)-stimulated cells by returning to this pathway [29][30][31][32][43,44,45,46]. Monocyte chemotactic protein-1-induced protein 1 (MCPIP1) inhibits macrophage polarization of M1 and promotes polarization of M2 by regulating the ATF3-AP1S2 signaling pathway, limiting the expression of pro-inflammatory cytokines in monocytes from patients with active inflammatory bowel disease (IBD). ATF3 can bind to the AP1S2 promoter and induce inflammation [33][47]. Postprandial triglyceride-rich lipoprotein (TGRL) lipolysis products (TL) induce the expression of pro-inflammatory factors in human brain microvascular endothelial cells by upregulating ATF3 through the activation of mitochondrial oxidative stress [34][48]. NF-κB regulates its expression by binding to the IL-6 promoter [35][36][49,50], while ATF3 has been shown to modulate NF-κB-dependent transcription by altering the phosphorylation of IκBα [22][34]. In osteoarthritis (OA), ATF3 modulates phosphorylation of p65 by altering IκB phosphorylation, which regulates the NF-κB signaling pathway, thus regulating the expression of IL-6 in chondrocytes [37][51]. ATF3 mediates particulate matter (PM)-induced inflammatory cytokine expression through the NF-κB and AP-1 pathways [38][52]. ATF3 plays a dual role in the regulation of inflammatory responses (Figure 1). In endotoxin-stimulated monocytes, the stress associated with reactive oxygen species (ROS) leads to the induction of ATF3 expression and inhibits IL-6 production, making mice highly susceptible to secondary bacterial and fungal infections [39][53]. Negative regulation of transcription by ATF3 may be achieved through the inhibition of CCAAT/enhancer binding protein δ (C/EBPδ), a positive regulator of cytokine gene induction [40][41][54,55]. C/EBPδ enhances the expression of IL-6 [40][54], and in turn, IL-6, through activation of IL-6R, can activate the STAT3, SHP-2, and MAPK pathways, thus promoting cytokine secretion [40][42][43][54,56,57]. It is reported that ATF3 recruits HDAC1 to the ATF3/p65 complex and promotes the deacetylation of p65 to inhibit the production of pro-inflammatory cytokines [44][58]. The role of ATF3 in macrophages is not limited to the regulation of pro-inflammatory cytokines. It is also essential for regulating the production of interferon (IFN)-β downstream of innate immune receptors. ATF3 acts as a transcriptional repressor by binding to regulatory sites on the Ifnb1 promoter [45][59]. The expression of macrophage inflammatory protein 1β (CCL4) leads to the onset of inflammatory diseases [30][46][44,60]. ATF3 inhibits the expression of CCL4 in mouse macrophages induced by LPS [30][44]. ATF3 negatively regulates the gene expression of IL-6 and IL-12 in macrophages by altering the structure of chromatin [13][29][19,43]. LPS activation of TLR4 induces ATF3 expression, which in turn suppresses the expression of various inflammatory genes induced by TLR4 signaling, including IL-6, IL-12b, and TNF-α [13][14][19,20].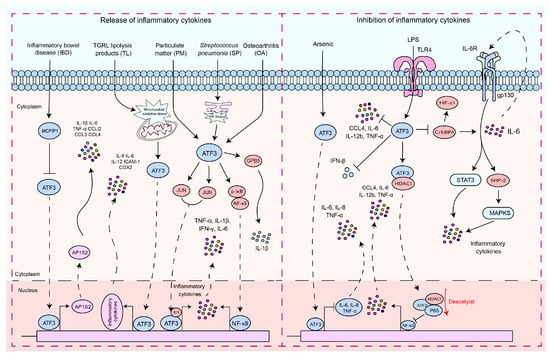
Figure 1. The signaling pathways by which ATF3 regulates the inflammatory response. These signaling pathways by which ATF3 regulates the inflammatory response can be divided into two types: (1) pro-inflammatory response—ATF3 increased the production of pro-inflammatory cytokines and chemokines by enhancing AP1S2 expression by binding to AP1S2 promoters in inflammation of the intestine (IBD). S. pneumoniae (SP) stimulates the formation of an ATF3 complex with c-Jun, and this complex binds to cytokine promoters of cytokines (TNF-α, IL-1β, and IFN-γ), resulting in increased cytokine production. During an infection, lung macrophages quickly phagocytose invasive S. pneumoniae, resulting in ER stress and ATF3 activation. ATF3 then promotes GBP5 activation, triggering IL-1β secretion. TGRL lipolysis products (TL) potentiate ROS in mitochondria, activating mitochondrial oxidative stress and ATF3 signaling. Furthermore, ATF3 regulates TL-induced inflammation. ATF3 may positively regulate IL-6 expression in osteoarthritis (OA) chondrocytes through modulation of NF-κB-dependent transcription by modifying IκB phosphorylation. ATF3 may heterodimerize with c-JUN and activate IL-6 transcription in HBE cells induced by PM. (2) Anti-inflammatory response, including regulation of the ATF3/HDAC1/NF-κB axis and ATF3/C-EBPδ axis—ATF3 inhibits the production of inflammatory cytokines by suppressing C/EBPδ. ATF3 inhibits the production of pro-inflammatory cytokines by recruiting HDAC1 into the ATF3/p65 complex and facilitating the deacetylation of p65. ATF3 acted as a transcriptional repressor and regulated IFN-β. LPS activates ATF3 by stimulating TLRs, thus inhibiting the production of inflammatory cytokines. Solid arrows indicate promotion, dashed arrows denote translocation, horizontal arrows represent inhibition, and red arrows signify the processes of modification undergone.
56. The Role of ATF3 in the Regulation of Cell Death
5.1. The Regulation of Apoptosis
6.1. The Regulation of Apoptosis
Apoptosis is a finely regulated process of programmed cell death that plays a crucial role in the maintenance of physiological functions in organisms, as well as in interactions between pathogens and hosts [47][48][49][50][63,64,65,66]. Apoptosis is a form of cell death by which the body maintains the homeostasis of the internal environment [5][51][5,67]. In addition, it plays an important role in the regulation of the immune system, especially in autoreactive immune cells [5][52][53][54][5,68,69,70]. In the interaction between pathogens and hosts, apoptosis can serve as a host defense mechanism to limit the replication and spread of pathogens [5][55][56][5,71,72]. In contrast, certain pathogens may exploit host defense mechanisms to evade host immune responses or induce excessive inflammation and tissue damage during infection [5][55][56][5,71,72]. Therefore, the diverse and complex functions of apoptosis are essential to understanding cellular behavior in physiological and pathological states. In particular, ATF3 induction appears to be consistently associated with cellular damage, as most of the signals that induce ATF3 also induce cellular injury [15][23][57][21,32,73]. Interestingly, ATF3 exhibits a dual role in the regulation of apoptosis (Figure 2). In cardiomyocytes, ATF3 effectively inhibits doxorubicin-induced apoptosis [58][74].
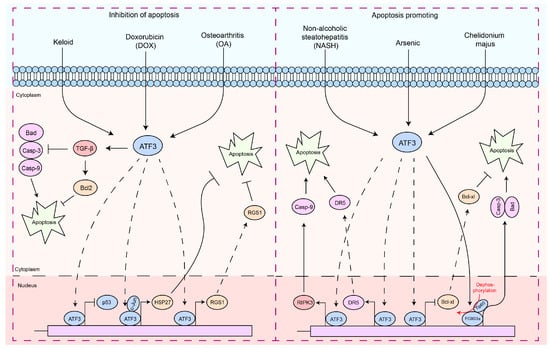
Figure 2. The functions of ATF3 in the response to apoptosis. These signaling pathways by which ATF3 regulates the apoptosis response can be divided into two types: (1) antiapoptotic response—ATF3 can regulate apoptosis of cells by upregulating the expression of the anti-apoptotic gene (HSP27, RGS1, and Bcl2) by binding to promoters, preventing p53 expression, which inhibits Caspases-3/9 activities. In addition, ATF3 inhibits Bad expression via TGF-β. (2) Pro-apoptosis response—ATF3 triggers the apoptotic pathway by upregulating RIPK3, DR5, and Caspase-9 by binding to their promoter, while concurrently inhibiting BCL-XL by binding to its promoter. Furthermore, ATF3 can also promote Caspase-3 and Bad transcription by activating FOXO3a, thereby regulating cell apoptosis. Solid arrows indicate promotion, dashed arrows denote translocation, horizontal arrows represent inhibition, and red arrows signify the processes of modification undergone.
These findings suggest a role for ATF3 in inhibiting apoptosis. However, ectopic expression of ATF3 improves the apoptotic capacity of topotecan-induced HeLa cells or camptothecin-induced HeLa cells [59][84]. ATF3 may act as a downstream target of the NF-κB and JNK/SAPK signaling pathways, promoting β-cell apoptosis [57][73]. ATF3 expression intensifies t-butyl hydroperoxide (TBHP)-induced apoptosis in nucleus pulposus cells (NPC) [60][85]. ATF3-dependent induction of RIPK3 causes a shift from apoptosis to necroptosis in hepatocytes [61][86]. The opposing regulation of DR5 and Bcl-xL expression by ATF3 promotes arsenic-induced apoptosis [62][87]. Forkhead transcription factors (FOXO3a) are a key molecule that promotes apoptosis, primarily functioning by facilitating the transcription of apoptosis-related factors, thereby mediating cell apoptosis [63][64][65][66][88,89,90,91]. The PI3K/Akt pathway inhibits apoptosis by phosphorylating FOXO3a, which prevents its nuclear translocation [65][66][67][90,91,92]. Chelidonium majus induces apoptosis in SKOV-3 cells by increasing the expression levels of ATF3 and its downstream protein, Tip60 [68][93].
5.2. The Regulation of Ferroptosis
6.2. The Regulation of Ferroptosis
Ferroptosis is a nonapoptotic form of cell death that can be induced by metabolic stress, such as glutathione (GSH) depletion [69][70][71][28,99,100]. Recently defined as a newly discovered form of cell death, ferroptosis is different from apoptosis in that it does not involve caspase activation [70][99]. Ferroptosis leads to an increase in ROS and malondialdehyde (MDA), which ultimately causes overwhelming lipid peroxidation and results in cell death [70][72][73][99,101,102]. ATF3 is often involved in vital cellular activities such as metabolism. Currently, a large amount of research data indicates that ATF3 plays a significant role in the regulation of ferroptosis [69][73][74][75][76][77][78][79][28,31,102,103,104,105,106,107]. Nuclear factor erythroid 2 related factor 2 (Nrf2) can promote the expression of SLC7A11 and GPX4 under oxidative stress, which is crucial to mediate the onset of ferroptosis [80][81][82][108,109,110]. As an endogenous inhibitor of SLC7A11, ATF3 promotes erastin-induced ferroptosis by inhibiting the cystine/glutamate antiporter (system Xc-) [69][28].