Gamma-aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the central nervous system (CNS). Most GABAergic neurons synthesize GABA from glutamate and release it in the synaptic cleft in the CNS. However, astrocytes can also synthesize and release GABA, activating GABA receptors in the neighboring neurons in physiological and pathological conditions. As the primary homeostatic glial cells in the brain, astrocytes play a crucial role in regulating GABA homeostasis and synaptic neurotransmission. Accumulating evidence demonstrates that astrocytic GABA dysregulation is implicated in psychiatric disorders, including alcohol use disorder (AUD) and major depressive disorder (MDD), the most prevalent co-occurring psychiatric disorders.
- GABA
- astrocyte
- alcohol use disorder
- major depressive disorder
- GABA transporter
- GABA receptor
1. Introduction
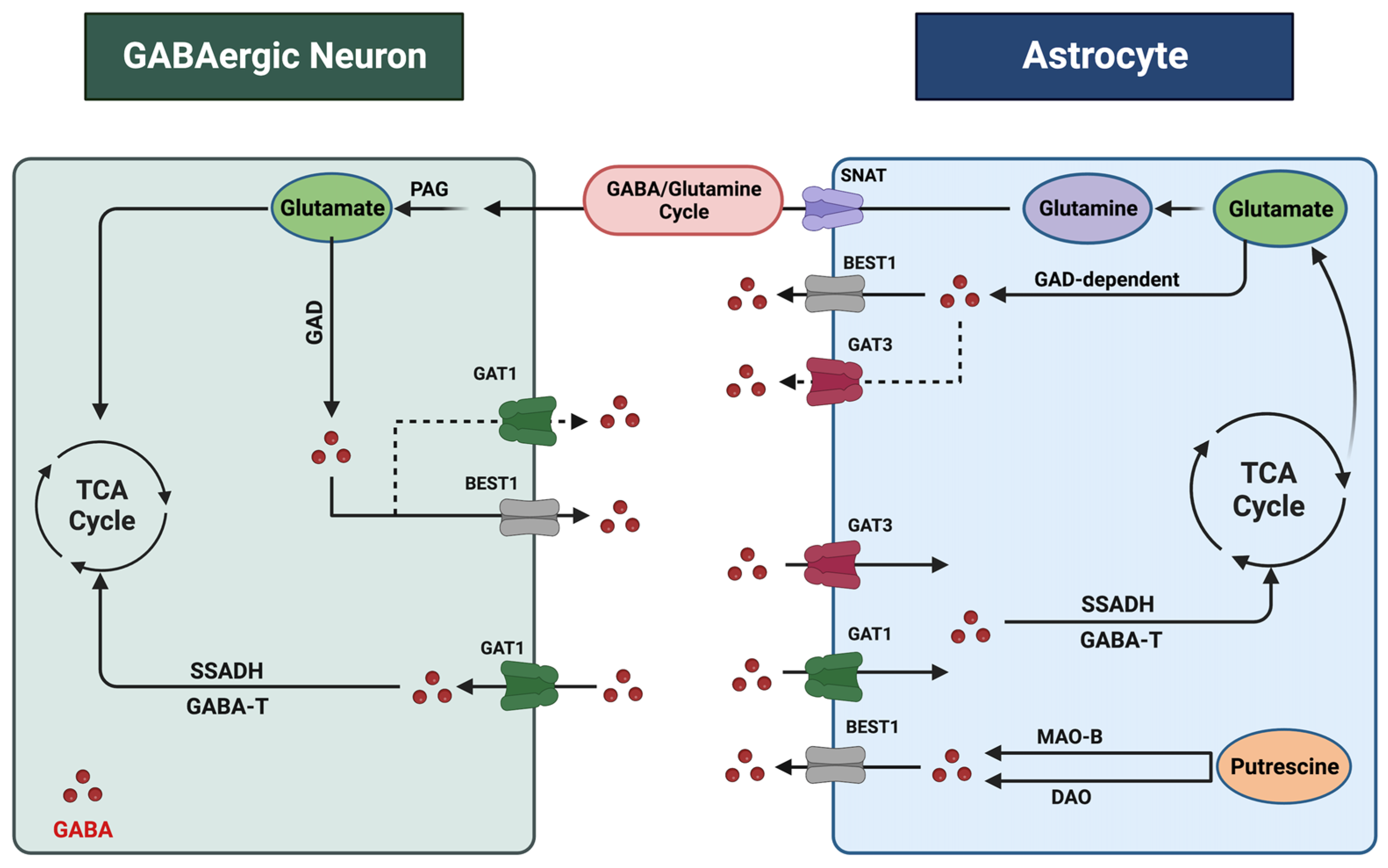
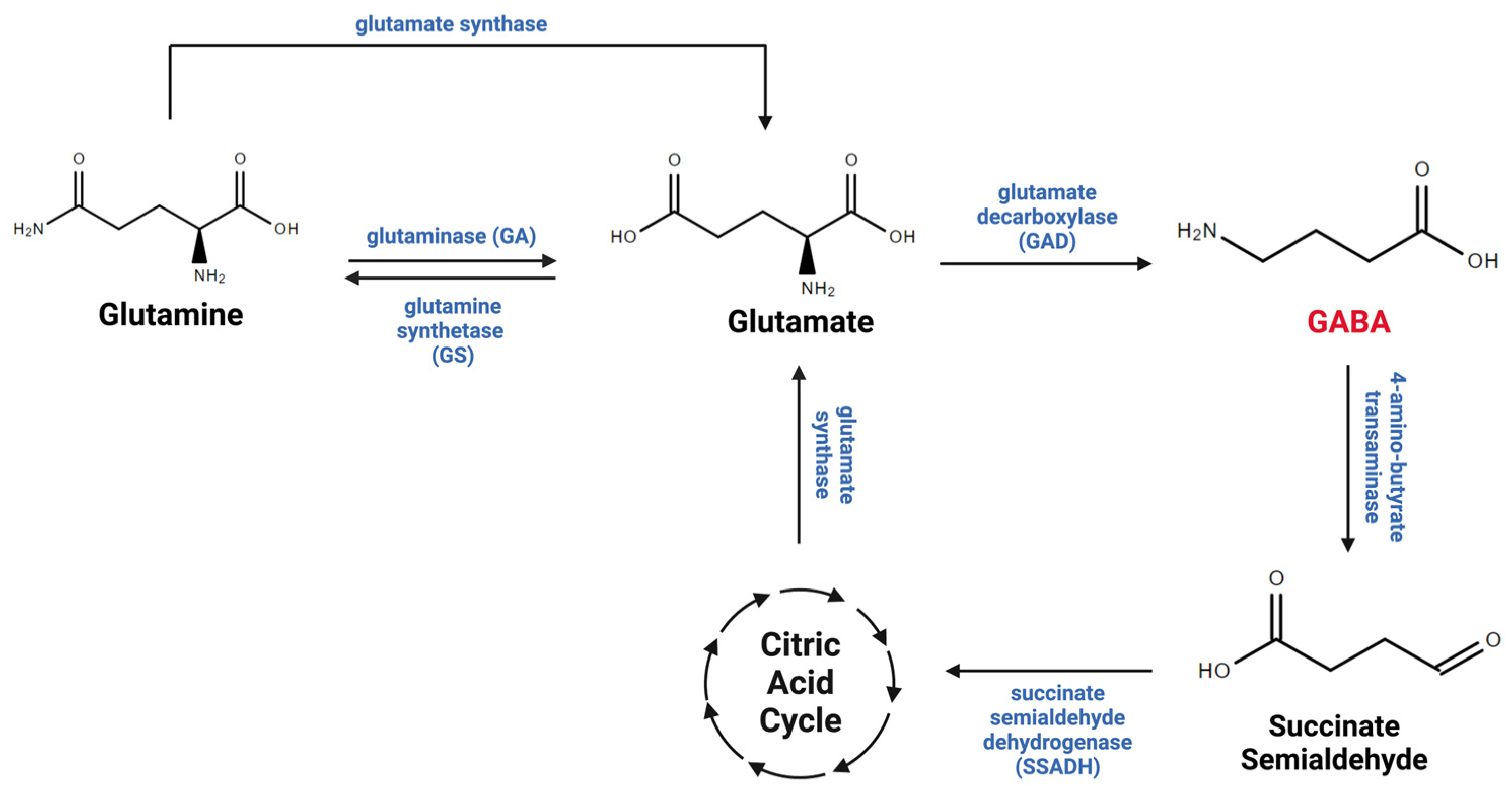
2. Astrocytic Regulation of Gamma-ABAminobutyric Acid in the CNentral Nervous System
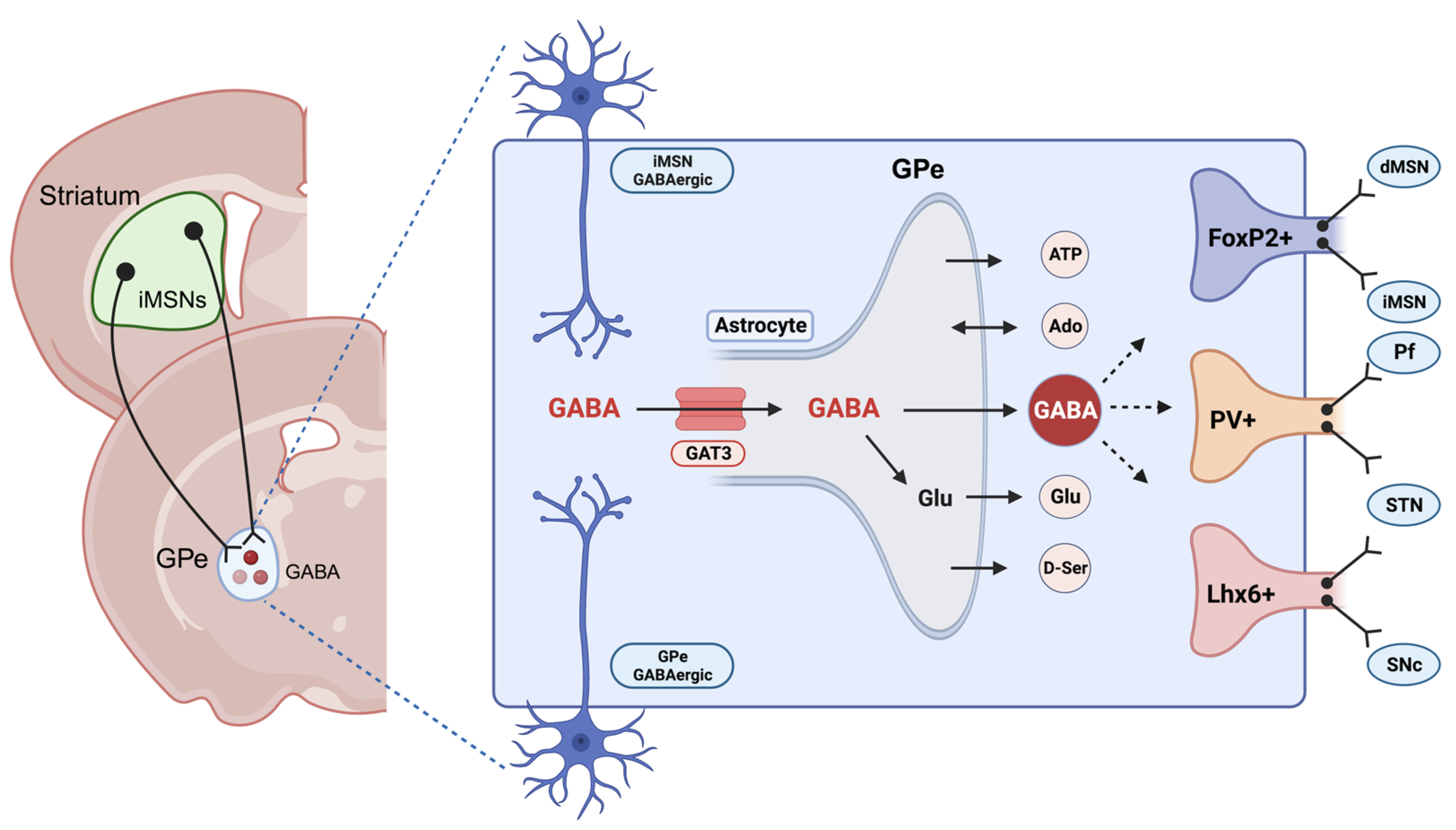
3. Astrocytes and Gamma-ABAminobutyric Acid in Alcohol Use Disorder (AUD)
References
- Whiteford, H.A.; Degenhardt, L.; Rehm, J.; Baxter, A.J.; Ferrari, A.J.; Erskine, H.E.; Charlson, F.J.; Norman, R.E.; Flaxman, A.D.; Johns, N. Global burden of disease attributable to mental and substance use disorders: Findings from the Global Burden of Disease Study 2010. Lancet 2013, 382, 1575–1586.
- Rodgers, B.; Korten, A.E.; Jorm, A.F.; Jacomb, P.A.; Christensen, H.; Henderson, A.S. Non-linear relationships in associations of depression and anxiety with alcohol use. Psychol. Med. 2000, 30, 421–432.
- Pedrelli, P.; Shapero, B.; Archibald, A.; Dale, C. Alcohol use and depression during adolescence and young adulthood: A summary and interpretation of mixed findings. Curr. Addict. Rep. 2016, 3, 91–97.
- Miguel-Hidalgo, J.J.; Wilson, B.A.; Hussain, S.; Meshram, A.; Rajkowska, G.; Stockmeier, C.A. Reduced connexin 43 immunolabeling in the orbitofrontal cortex in alcohol dependence and depression. J. Psychiatr. Res. 2014, 55, 101–109.
- Miguel-Hidalgo, J.J.; Waltzer, R.; Whittom, A.A.; Austin, M.C.; Rajkowska, G.; Stockmeier, C.A. Glial and glutamatergic markers in depression, alcoholism, and their comorbidity. J. Affect. Disord. 2010, 127, 230–240.
- Brière, F.N.; Rohde, P.; Seeley, J.R.; Klein, D.; Lewinsohn, P.M. Comorbidity between major depression and alcohol use disorder from adolescence to adulthood. Compr. Psychiatry 2014, 55, 526–533.
- Cornelius, J.R.; Bukstein, O.; Salloum, I.; Clark, D. Alcohol and psychiatric comorbidity. Recent. Dev. Alcohol. Res. Alcohol. Treat. 2002, 16, 361–374.
- Grant, B.F.; Harford, T.C. Comorbidity between DSM-IV alcohol use disorders and major depression: Results of a national survey. Drug Alcohol. Depend. 1995, 39, 197–206.
- Neupane, S.P. Neuroimmune interface in the comorbidity between alcohol use disorder and major depression. Front. Immunol. 2016, 7, 655.
- Khalid, A.; Kunwar, A.R.; Rajbhandari, K.; Sharma, V.D.; Regmi, S.K. A study of prevalence and comorbidity of depression in alcohol dependence. Indian. J. Psychiatry 2000, 42, 434.
- Currie, S.R.; Patten, S.B.; Williams, J.V.; Wang, J.; Beck, C.A.; El-Guebaly, N.; Maxwell, C. Comorbidity of major depression with substance use disorders. Can. J. Psychiatry 2005, 50, 660–666.
- Luscher, B.; Shen, Q.; Sahir, N. The GABAergic deficit hypothesis of major depressive disorder. Mol. Psychiatry 2011, 16, 383–406.
- Koob, G.F. A role for GABA mechanisms in the motivational effects of alcohol. Biochem. Pharmacol. 2004, 68, 1515–1525.
- Koob, G.F. A role for GABA in alcohol dependence1. Adv. Pharmacol. 2006, 54, 205–229.
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896.
- Zurek, A.A.; Yu, J.; Wang, D.-S.; Haffey, S.C.; Bridgwater, E.M.; Penna, A.; Lecker, I.; Lei, G.; Chang, T.; Salter, E.W. Sustained increase in α5GABA A receptor function impairs memory after anesthesia. J. Clin. Investig. 2014, 124, 5437–5441.
- Woo, J.; Min, J.O.; Kang, D.-S.; Kim, Y.S.; Jung, G.H.; Park, H.J.; Kim, S.; An, H.; Kwon, J.; Kim, J. Control of motor coordination by astrocytic tonic GABA release through modulation of excitation/inhibition balance in cerebellum. Proc. Natl. Acad. Sci. USA 2018, 115, 5004–5009.
- Marowsky, A.; Rudolph, U.; Fritschy, J.-M.; Arand, M. Tonic inhibition in principal cells of the amygdala: A central role for α3 subunit-containing GABAA receptors. J. Neurosci. 2012, 32, 8611–8619.
- Kwak, H.; Koh, W.; Kim, S.; Song, K.; Shin, J.-I.; Lee, J.M.; Lee, E.H.; Bae, J.Y.; Ha, G.E.; Oh, J.-E. Astrocytes control sensory acuity via tonic inhibition in the thalamus. Neuron 2020, 108, 691–706.e10.
- Olsen, R.W.; Hanchar, H.J.; Meera, P.; Wallner, M. GABAA receptor subtypes: The “one glass of wine” receptors. Alcohol 2007, 41, 201–209.
- Weyand, T.G.; Boudreaux, M.; Guido, W. Burst and tonic response modes in thalamic neurons during sleep and wakefulness. J. Neurophysiol. 2001, 85, 1107–1118.
- Fogaça, M.V.; Duman, R.S. Cortical GABAergic Dysfunction in Stress and Depression: New Insights for Therapeutic Interventions. Front. Cell. Neurosci. 2019, 13, 87.
- Roberto, M.; Kirson, D.; Khom, S. The Role of the Central Amygdala in Alcohol Dependence. Cold Spring Harb. Perspect. Med. 2021, 11, a039339.
- Liljequist, S.; Engel, J. Effects of GABAergic agonists and antagonists on various ethanol-induced behavioral changes. Psychopharmacology 1982, 78, 71–75.
- Vengeliene, V.; Bilbao, A.; Molander, A.; Spanagel, R. Neuropharmacology of alcohol addiction. Br. J. Pharmacol. 2008, 154, 299–315.
- Cousins, M.S.; Roberts, D.C.; de Wit, H. GABA(B) receptor agonists for the treatment of drug addiction: A review of recent findings. Drug Alcohol. Depend. 2002, 65, 209–220.
- Janak, P.H.; Michael Gill, T. Comparison of the effects of allopregnanolone with direct GABAergic agonists on ethanol self-administration with and without concurrently available sucrose. Alcohol 2003, 30, 1–7.
- Grant, K.A.; Waters, C.A.; Green-Jordan, K.; Azarov, A.; Szeliga, K.T. Characterization of the discriminative stimulus effects of GABA(A) receptor ligands in Macaca fascicularis monkeys under different ethanol training conditions. Psychopharmacology 2000, 152, 181–188.
- Rassnick, S.; D’Amico, E.; Riley, E.; Koob, G.F. GABA antagonist and benzodiazepine partial inverse agonist reduce motivated responding for ethanol. Alcohol. Clin. Exp. Res. 1993, 17, 124–130.
- Jazvinscak Jembrek, M.; Vlainic, J. GABA receptors: Pharmacological potential and pitfalls. Curr. Pharm. Des. 2015, 21, 4943–4959.
- Sigel, E.; Steinmann, M.E. Structure, function, and modulation of GABAA receptors. J. Biol. Chem. 2012, 287, 40224–40231.
- Watanabe, M.; Maemura, K.; Kanbara, K.; Tamayama, T.; Hayasaki, H. GABA and GABA receptors in the central nervous system and other organs. Int. Rev. Cytol. 2002, 213, 1–47.
- Pinard, A.; Seddik, R.; Bettler, B. GABAB receptors: Physiological functions and mechanisms of diversity. Adv. Pharmacol. 2010, 58, 231–255.
- Lee, M.; Schwab, C.; Mcgeer, P.L. Astrocytes are GABAergic cells that modulate microglial activity. Glia 2011, 59, 152–165.
- Liu, J.; Feng, X.; Wang, Y.; Xia, X.; Zheng, J.C. Astrocytes: GABAceptive and GABAergic cells in the brain. Front. Cell. Neurosci. 2022, 16, 892497.
- Andersen, J.V.; Schousboe, A.; Wellendorph, P. Astrocytes regulate inhibitory neurotransmission through GABA uptake, metabolism, and recycling. Essays Biochem. 2023, 67, 77–91.
- Kilb, W.; Kirischuk, S. GABA release from astrocytes in health and disease. Int. J. Mol. Sci. 2022, 23, 15859.
- Lee, M.; McGeer, E.G.; McGeer, P.L. Mechanisms of GABA release from human astrocytes. Glia 2011, 59, 1600–1611.
- Schousboe, A.; Hertz, L.; Svenneby, G. Uptake and metabolism of GABA in astrocytes cultured from dissociated mouse brain hemispheres. Neurochem. Res. 1977, 2, 217–229.
- Schousboe, A.; Waagepetersen, H.S. Role of astrocytes in homeostasis of glutamate and GABA during physiological and pathophysiological conditions. Adv. Mol. Cell Biol. 2003, 31, 461–474.
- Schousboe, A.; Westergaard, N.; Sonnewald, U.; Petersen, S.; Yu, A.; Hertz, L. Regulatory role of astrocytes for neuronal biosynthesis and homeostasis of glutamate and GABA. Prog. Brain Res. 1992, 94, 199–211.
- Ishibashi, M.; Egawa, K.; Fukuda, A. Diverse Actions of Astrocytes in GABAergic Signaling. Int. J. Mol. Sci. 2019, 20, 2964.
- Roberts, E.; Frankel, S. γ-Aminobutyric acid in brain: Its formation from glutamic acid. J. Biol. Chem. 1950, 187, 55–63.
- Tao, R.; Davis, K.N.; Li, C.; Shin, J.H.; Gao, Y.; Jaffe, A.E.; Gondré-Lewis, M.C.; Weinberger, D.R.; Kleinman, J.E.; Hyde, T.M. GAD1 alternative transcripts and DNA methylation in human prefrontal cortex and hippocampus in brain development, schizophrenia. Mol. Psychiatry 2018, 23, 1496–1505.
- Yoon, B.E.; Woo, J.; Chun, Y.E.; Chun, H.; Jo, S.; Bae, J.Y.; An, H.; Min, J.O.; Oh, S.J.; Han, K.S. Glial GABA, synthesized by monoamine oxidase B, mediates tonic inhibition. J. Physiol. 2014, 592, 4951–4968.
- Losi, G.; Mariotti, L.; Carmignoto, G. GABAergic interneuron to astrocyte signalling: A neglected form of cell communication in the brain. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130609.
- Scimemi, A. Structure, function, and plasticity of GABA transporters. Front. Cell. Neurosci. 2014, 8, 161.
- Jin, X.T.; Galvan, A.; Wichmann, T.; Smith, Y. Localization and Function of GABA Transporters GAT-1 and GAT-3 in the Basal Ganglia. Front. Syst. Neurosci. 2011, 5, 63.
- Fattorini, G.; Catalano, M.; Melone, M.; Serpe, C.; Bassi, S.; Limatola, C.; Conti, F. Microglial expression of GAT-1 in the cerebral cortex. Glia 2020, 68, 646–655.
- Melone, M.; Ciappelloni, S.; Conti, F. A quantitative analysis of cellular and synaptic localization of GAT-1 and GAT-3 in rat neocortex. Brain Struct. Funct. 2015, 220, 885–897.
- Minelli, A.; DeBiasi, S.; Brecha, N.C.; Zuccarello, L.V.; Conti, F. GAT-3, a high-affinity GABA plasma membrane transporter, is localized to astrocytic processes, and it is not confined to the vicinity of GABAergic synapses in the cerebral cortex. J. Neurosci. 1996, 16, 6255–6264.
- Boddum, K.; Jensen, T.P.; Magloire, V.; Kristiansen, U.; Rusakov, D.A.; Pavlov, I.; Walker, M.C. Astrocytic GABA transporter activity modulates excitatory neurotransmission. Nat. Commun. 2016, 7, 13572.
- Andersen, J.V.; Schousboe, A. Milestone Review: Metabolic dynamics of glutamate and GABA mediated neurotransmission—The essential roles of astrocytes. J. Neurochem. 2023, 166, 109–137.
- Tritsch, N.X.; Granger, A.J.; Sabatini, B.L. Mechanisms and functions of GABA co-release. Nat. Rev. Neurosci. 2016, 17, 139–145.
- Murphy-Royal, C.; Ching, S.; Papouin, T. A conceptual framework for astrocyte function. Nat. Neurosci. 2023, 26, 1848–1856.
- Waagepetersen, H.S.; Sonnewald, U.; Schousboe, A. Compartmentation of glutamine, glutamate, and GABA metabolism in neurons and astrocytes: Functional implications. Neurosci. 2003, 9, 398–403.
- Carvalho, A.F.; Heilig, M.; Perez, A.; Probst, C.; Rehm, J. Alcohol use disorders. Lancet 2019, 394, 781–792.
- Lindberg, D.; Andres-Beck, L.; Jia, Y.F.; Kang, S.; Choi, D.S. Purinergic Signaling in Neuron-Astrocyte Interactions, Circadian Rhythms, and Alcohol Use Disorder. Front. Physiol. 2018, 9, 9.
- Yang, W.; Singla, R.; Maheshwari, O.; Fontaine, C.J.; Gil-Mohapel, J. Alcohol Use Disorder: Neurobiology and Therapeutics. Biomedicines 2022, 10, 1192.
- Griswold, M.G.; Fullman, N.; Hawley, C.; Arian, N.; Zimsen, S.R.; Tymeson, H.D.; Venkateswaran, V.; Tapp, A.D.; Forouzanfar, M.H.; Salama, J.S. Alcohol use and burden for 195 countries and territories, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2018, 392, 1015–1035.
- World Health Organization. Global Status Report on Alcohol and Health 2018; World Health Organization: Geneva, Switzerland, 2019.
- Collaborators, G.; Ärnlöv, J. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1223–1249.
- Koob, G.F.; Roberts, A.J.; Schulteis, G.; Parsons, L.H.; Heyser, C.J.; Hyytiä, P.; Merlo-Pich, E.; Weiss, F. Neurocircuitry targets in ethanol reward and dependence. Alcohol. Clin. Exp. Res. 1998, 22, 3–9.
- Augier, E.; Barbier, E.; Dulman, R.S.; Licheri, V.; Augier, G.; Domi, E.; Barchiesi, R.; Farris, S.; Nätt, D.; Mayfield, R.D.; et al. A molecular mechanism for choosing alcohol over an alternative reward. Science 2018, 360, 1321–1326.
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016, 3, 760–773.
- Domi, E.; Domi, A.; Adermark, L.; Heilig, M.; Augier, E. Neurobiology of alcohol seeking behavior. J. Neurochem. 2021, 157, 1585–1614.
- Abrahao, K.P.; Salinas, A.G.; Lovinger, D.M. Alcohol and the brain: Neuronal molecular targets, synapses, and circuits. Neuron 2017, 96, 1223–1238.
- Koob, G.F.; Volkow, N.D. Neurocircuitry of addiction. Neuropsychopharmacology 2010, 35, 217–238.
- Spanagel, R.; Zink, M.; Sommer, W.H. Neurobiology of Alcohol Addiction. In Neuroscience in the 21st Century: From Basic to Clinical; Pfaff, D.W., Ed.; Springer: New York, NY, USA, 2013; pp. 2745–2773.
- Roberto, M.; Madamba, S.G.; Stouffer, D.G.; Parsons, L.H.; Siggins, G.R. Increased GABA release in the central amygdala of ethanol-dependent rats. J. Neurosci. 2004, 24, 10159–10166.
- Colombo, G.; Serra, S.; Brunetti, G.; Vacca, G.; Carai, M.A.; Gessa, G.L. Suppression by baclofen of alcohol deprivation effect in Sardinian alcohol-preferring (sP) rats. Drug Alcohol. Depend. 2003, 70, 105–108.
- GOLDSTEIN, D.B. Alcohol withdrawal reactions in mice: Effects of drugs that modify neurotransmission. J. Pharmacol. Exp. Ther. 1973, 186, 1–9.
- Hyytiä, P.; Koob, G.F. GABAA receptor antagonism in the extended amygdala decreases ethanol self-administration in rats. Eur. J. Pharmacol. 1995, 283, 151–159.
- Foster, K.L.; McKay, P.F.; Seyoum, R.; Milbourne, D.; Yin, W.; Sarma, P.; Cook, J.M.; June, H.L. GABAA and opioid receptors of the central nucleus of the amygdala selectively regulate ethanol-maintained behaviors. Neuropsychopharmacology 2004, 29, 269–284.
- Frye, G.D.; Mccown, T.J.; Breese, G.R. Characterization of susceptibility to audiogenic seizures in ethanol-dependent rats after microinjection of γ-aminobutyric acid (GABA) agonists into the inferior colliculus, substantia nigra or medial septum. J. Pharmacol. Exp. Ther. 1983, 227, 663.
- Cooper, B.; Viik, K.; Ferris, R.; White, H. Antagonism of the enhanced susceptibility to audiogenic seizures during alcohol withdrawal in the rat by gamma-aminobutyric acid (GABA) and” GABA-mimetic” agents. J. Pharmacol. Exp. Ther. 1979, 209, 396–403.
- Frye, G.D.; McCOWN, T.J.; Breese, G.R. Differential sensitivity of ethanol withdrawal signs in the rat to gamma-aminobutyric acid (GABA) mimetics: Blockade of audiogenic seizures but not forelimb tremors. J. Pharmacol. Exp. Ther. 1983, 226, 720–725.
- Valenzuela, C.F. Alcohol and neurotransmitter interactions. Alcohol. Health Res. World 1997, 21, 144–148.
- Mederos, S.; Perea, G. GABAergic-astrocyte signaling: A refinement of inhibitory brain networks. Glia 2019, 67, 1842–1851.
- Corbit, L.H.; Nie, H.; Janak, P.H. Habitual alcohol seeking: Time course and the contribution of subregions of the dorsal striatum. Biol. Psychiatry 2012, 72, 389–395.
- Barker, J.M.; Taylor, J.R. Habitual alcohol seeking: Modeling the transition from casual drinking to addiction. Neurosci. Biobehav. Rev. 2014, 47, 281–294.
- Olsen, R.W.; Liang, J. Role of GABAA receptors in alcohol use disorders suggested by chronic intermittent ethanol (CIE) rodent model. Mol. Brain 2017, 10, 45.
- Lovinger, D.M.; Kash, T.L. Mechanisms of neuroplasticity and ethanol’s effects on plasticity in the striatum and bed nucleus of the stria terminalis. Alcohol Res. Curr. Rev. 2015, 37, 109.
- Mederos, S.; Sánchez-Puelles, C.; Esparza, J.; Valero, M.; Ponomarenko, A.; Perea, G. GABAergic signaling to astrocytes in the prefrontal cortex sustains goal-directed behaviors. Nat. Neurosci. 2021, 24, 82–92.
- Lipton, D.M.; Gonzales, B.J.; Citri, A. Dorsal Striatal Circuits for Habits, Compulsions and Addictions. Front. Syst. Neurosci. 2019, 13, 28.
- Yin, H.H.; Knowlton, B.J. The role of the basal ganglia in habit formation. Nat. Rev. Neurosci. 2006, 7, 464–476.
- Kang, S.; Choi, D.S. Astrocyte adenosine signaling and neural mechanisms of goal-directed and habitual reward-seeking behaviors. Neuropsychopharmacology 2021, 46, 227–228.
- Mallet, N.; Micklem, B.R.; Henny, P.; Brown, M.T.; Williams, C.; Bolam, J.P.; Nakamura, K.C.; Magill, P.J. Dichotomous organization of the external globus pallidus. Neuron 2012, 74, 1075–1086.
- Hong, S.-I.; Kang, S.; Song, M.; Yang, M.; Baker, M.; Kang, S.; Lee, J.; Lee, S.W.; Choi, D.-S. Astrocytes in the external globus pallidus coordinate flexibility of action strategy. Sci. Adv. 2021, 9, eadh9239.
- Hong, S.I.; Kang, S.; Baker, M.; Choi, D.S. Astrocyte-neuron interaction in the dorsal striatum-pallidal circuits and alcohol-seeking behaviors. Neuropharmacology 2021, 198, 108759.
- Kang, S.; Hong, S.I.; Kang, S.; Song, M.; Yang, M.A.; Essa, H.; Baker, M.; Lee, J.; Bruce, R.A.; Lee, S.W.; et al. Astrocyte activities in the external globus pallidus regulate action-selection strategies in reward-seeking behaviors. Sci. Adv. 2023, 9, eadh9239.
- Chazalon, M.; Paredes-Rodriguez, E.; Morin, S.; Martinez, A.; Cristóvão-Ferreira, S.; Vaz, S.; Sebastiao, A.; Panatier, A.; Boué-Grabot, E.; Miguelez, C. GAT-3 dysfunction generates tonic inhibition in external globus pallidus neurons in parkinsonian rodents. Cell Rep. 2018, 23, 1678–1690.
- Ribak, C.E.; Tong, W.M.; Brecha, N.C. GABA plasma membrane transporters, GAT-1 and GAT-3, display different distributions in the rat hippocampus. J. Comp. Neurol. 1996, 367, 595–606.
- Morrow, A.L.; Suzdak, P.D.; Karanian, J.W.; Paul, S.M. Chronic ethanol administration alters gamma-aminobutyric acid, pentobarbital and ethanol-mediated 36Cl- uptake in cerebral cortical synaptoneurosomes. J. Pharmacol. Exp. Ther. 1988, 246, 158–164.
- Szmigielski, A.; Szmigielska, H.; Wejman, I. The effect of single and prolonged ethanol administration on the sensitivity of central GABA-A and benzodiazepine receptors in vivo. Pol. J. Pharmacol. Pharm. 1992, 44, 271–280.
- Roberts, A.J.; Cole, M.; Koob, G.F. Intra-amygdala muscimol decreases operant ethanol self-administration in dependent rats. Alcohol. Clin. Exp. Res. 1996, 20, 1289–1298.
- Marti-Prats, L.; Belin-Rauscent, A.; Fouyssac, M.; Puaud, M.; Cocker, P.J.; Everitt, B.J.; Belin, D. Baclofen decreases compulsive alcohol drinking in rats characterized by reduced levels of GAT-3 in the central amygdala. Addict. Biol. 2021, 26, e13011.