2. The HIV-1 Tat Protein
Tat is a 14–16 kD HIV regulatory protein whose main role in the HIV life cycle is to promote virus transcription, and primarily transcript elongation. In fact, Tat is prominently known for its role in relieving RNA polymerase II from pause, thus promoting elongation, a key step leading to the completion of HIV gene transcription
[14]. However, Tat is also required to initiate reverse transcription (RT)
[7], to increase the rate of transcription
[7] and to contribute to splicing regulation
[15][16][15,16].
Tat is generated in two forms through alternative splicing. The first form is encoded by the multiply spliced two-exon transcript and varies in length between 86 and 101 amino acids, depending on the viral isolate, whereas the other form is encoded by a singly spliced one-exon transcript and is 72 amino acids long. Both Tat variants transactivate the LTR efficiently, but the two-exon Tat appears to exert additional effects on the infected cell, such as altering cytoskeleton structure and function
[17], delaying Fas-mediated apoptosis
[18], and reducing the triggering of innate and adaptive immune responses by downregulating expression of interferon-stimulated genes and MHC class-I and II molecules in antigen-presenting cells
[19][20][21][19,20,21]. Here, unless differently stated, Tat refers to the 86 amino acids (aa)-long two-exon Tat protein, which is the most commonly used form of Tat
[22].
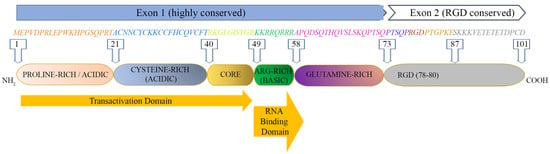
The Tat protein is largely unstructured and contains six functional domains (
Figure 1). These features make Tat capable of interacting with many molecules, as it can easily adapt to a molecule displaying one or more complementary domains or motifs.
Figure 1.
HIV-1 Tat sequence (HXBc2 strain) and functional domains. See text for Tat domains’ detailed description.
The first domain (1–21 aa) at the N-terminus is an acidic proline-rich region and has a conserved Trp 11 that is critical to stabilize Tat binding to the inner leaf of the cell membrane
[12]; the second domain (residues 22–37) has seven cysteines that are rather well conserved at positions 22, 25, 27, 30, 31, 34 and 37; six of them may establish intramolecular bonds, while the seventh cysteine is believed to mediate intermolecular bridging; the third domain (core, residues 38–48) contains two residues of a conserved four-amino-acid subdomain (36–39) reported to bind tubulin/microtubules through a microtubule-associated protein, LIS1
[23], leading to the alteration of microtubule dynamics and activation of a mitochondria-dependent apoptotic pathway
[24][25][24,25]. Overall, the N-terminal half (1–48 aa, region I–III) of Tat is highly conserved, consistent with the critical role for activating the transcription of HIV genomic DNA due to the cysteine-rich motif (region II), required for dimerization, protein structure stabilization and metal binding, and the hydrophobic core motif (region III) required for binding to the CDK9-associated C-type cyclin
[26] and to the transactivation response RNA element (TAR) of the newly transcribed HIV genomic RNA
[27][28][27,28].
The C-terminal half (49–72 aa) of Tat contains two more regions: region IV (residues 49–55) is a conserved basic domain important for localizing Tat to the nucleus, binding of Tat to TAR and the internalization of the Tat protein into bystander cells by its interaction with surface proteins such as heparan sulfate proteoglycans (HSPG)
[29][30][29,30]. Of note, a peptide corresponding to the basic region is currently exploited as a penetratin or transducing molecule to intracellularly deliver numerous cargos (proteins, DNA, RNA), underscoring its ability to cross membranes
[31]. Residues 59–72 encompass the glutamine-rich domain (region V), which is less conserved and participates in the interaction of Tat with TAR and in Tat-mediated apoptosis
[32].
Although more variable than Exon 1, most Exon 2 sequences (region VI) contain an arginine-glycine-aspartic acid (RGD) motif (78–80 aa) which is important for the Tat interaction with the RGD-binding integrins αvβ1, αvβ3, α5β1
[33][34][33,34]. Whether and to what extent Tat binds the other 5 RGD binding integrins remains to be investigated. Although not required, the second exon of Tat contributes to optimal virus replication in T cells and macrophages
[35][36][35,36]. In T cells, Tat101 delayed FasL-mediated apoptosis, prolonging HIV-1 replication
[18], and altered the function and distribution of mitochondria
[37]. In myeloid cells, the two-exon protein has been reported to reduce the innate immunity activation observed with one-exon Tat
[19].
Tat is expressed very early upon infection, even before virus integration
[38]. Indeed, accumulating evidence indicates that Tat is incorporated into HIV-1 virions
[39][40][39,40], thus priming both intra-virion and post-entry reverse transcription
[41] and activating virus gene expression even prior to HIV gene expression
[40].
3. Mechanism and Kinetics of the Extracellular Release of Tat (eTat)
The majority (about 65%) of the Tat protein produced by the infected cell is released extracellularly in the absence of cell death or cell permeability changes mainly by a leaderless secretory pathway similar to that used by basic fibroblast growth factor (FGF)2 to exit cells
[8][10][11][12][42][8,10,11,12,42]. However, none of the unconventional secretory pathways used by FGF or interleukin (IL)-1 β are involved, as Tat appears to traffic to the plasma membrane without the contribution of intracellular intermediates
[12]. In particular, the conserved Arg49, Lys50 and Lys51 (RKK motif) of Tat appears to bind the fraction of phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2] located at the plasma membrane
[12]. This binding is strengthened by the insertion of Tat Trp11 into a hydrophobic cleft of the inner membrane
[12]. An additional interaction between the basic region (RRQRRR) of Tat and phosphatidylserine has also been reported very recently
[43]. These interactions may affect several biological processes in which PI(4,5)P2 is involved, such as clathrin-mediated endocytosis
[44], phagocytosis
[45][46][45,46] or exocytosis
[47]. Plasma-membrane-bound Tat is then released extracellularly by exocytosis
[11], with still incompletely understood mechanism(s) (reviewed in
[42]). Recent evidence indicates that Tat is also released extracellularly within exosomes, which appear to be enriched with small noncoding RNAs containing TAR and their derivative TAR miRNA
[48], which, in turn, have been reported to promote, respectively, inflammation and tumorigenesis, two hallmarks of the residual disease observed in subjects on long-term suppressive cART
[49][50][51][49,50,51]. Of note, extracellular Tat (eTat) crosses the blood–brain barrier and promotes the central nervous system (CNS) inflammation and T-cell activation
[52], which persist despite treatment intensification with CNS-penetrating ART
[53].
Upon release, eTat binds heparan sulphate proteoglycans (HSPG) of the extracellular matrix (ECM) and is detected in the tissues of infected individuals
[8][9][11][52][54][55][56][57][8,9,11,52,54,55,56,57]. eTat is biologically active and exerts activities key for the acquisition of infection, virus reactivation and HIV disease maintenance in cART-treated individuals
[9][10][11][12][38][55][56][57][58][59][60][61][62][63][64][65][66][67][68][9,10,11,12,38,55,56,57,58,59,60,61,62,63,64,65,66,67,68].
Up to 40 ng/mL (4 nM) of eTat has been detected in biological fluids, but these amounts are probably underestimated, as eTat accumulates in tissues, bound to the HSPG of the extracellular matrix (ECM) in a biologically active form
[8][52][54][55][56][57][8,52,54,55,56,57]. eTat is also known to be taken up by uninfected cells, including T lymphocytes, macrophages and neurosecretory cells and to accumulate at the plasma membrane, where it stably binds to phosphatidylinositol (4,5) bisphosphate (PI(4,5)P2), thus interfering with the (PI(4,5)P2)-mediated functions, such as neurosecretion, phagocytosis and cardiac muscle repolarization
[69]. For stable binding to (PI(4,5)P2), interaction of the Tatcys31 with cyclophilin A (CypA) and Tat palmitoylation by HHD20 are required
[69]. Intriguingly, the Indian clade C Tat displays a naturally occurring cysteine 31 substitution that abolishes (PI(4,5)P2) sequestration by Tat, possibly contributing to the apparent lower pathogenicity of clade C Tat as compared to clade B
[69]. In sharp contrast, Tat is efficiently released in productively infected cells. CypA binds the newly produced HIV-1 Gag protein, it is incorporated into the capsid and it is released from the cell within the budding virus
[40]. As a result, intracellular CypA is depleted and Tat palmitoylation cannot occur
[69].
4. Role of eTat in HIV Acquisition, Dissemination and Reservoir Formation
4.1. HIV Acquisition
HIV is mostly acquired through sexual intercourse, with the rate of acquisition being extremely low, which varies according to the type of unprotected sex (vaginal/anal/oral; insertive/receptive
[70][71][72][73][70,71,72,73], and it is strongly favored by pre-existing genital infections
[74] and viral load
[75][76][77][78][75,76,77,78]. Thus, mucosae represent a strong barrier to the acquisition of HIV (reviewed in
[79]), which must undergo a major selection to overcome the mucosal barrier
[80], resulting in the systemic spreading of founder viruses characterized by Env with a “short” V1/V2 loop
[81].
In this context,
theour data indicate that Tat binds native trimeric Env on HIV-1 virions to form a novel cell entry complex (the Tat/Env complex), enabling the virus to enter dendritic cells (DCs) through an integrin-mediated endocytic pathway alternative to the canonical endocytic pathway mediated by C-type lectin receptors
[82]. Tat-mediated entry in DCs leads to the enhancement of HIV infection in these cells
[82], which are key for HIV acquisition at the mucosal portal of entry. The Env V3 loop is a main Tat binding determinant, as a cyclic (but not linear) peptide encompassing the V3 loop was shown to bind recombinant Tat, and V3 loop deletion abrogates the capability of Tat to direct Env on the integrin receptors
[82]. In this regard, the Env V1/V2 loop is known to engage and occlude the V3 loop at the Env trimer apex
[83], and
our studies indicate that V1/V2 loop shortening dramatically increases the stability of the Tat/Env complex
[82]. These data might explain why V1/V2 shortening is a common feature of the founder viruses emerging at the mucosal portal of entry.
Since the Tat/Env complex drives HIV to the DCs integrin receptors, anti-Env Abs blocking the Env interaction with C-type lectin receptors become ineffective at preventing HIV-1 entry in these cells
[82]. However, entry is blocked by the addition of anti-Tat Abs
[82]. Indeed, the immunization of monkeys with the Tat/Env complex led to infection containment upon intrarectal challenge with a pathogenic simian-HIV chimeric virus (SHIV), preventing virus spreading beyond the rectum
[82]. Taken together, these data suggest that eTat plays a key role also in HIV-1 acquisition, providing the rationale to also include it in preventative vaccine approaches in association with Env.
4.2. HIV Dissemination
As Tat is required for HIV gene expression, it is apparent it plays a critical role in promoting virus replication and, ultimately, dissemination. Indeed, seroreversion of the antibody response to HIV, a bona fide proxy of virus remission or eradication, was reported in a small cohort of 23 women from Gabon infected with a Tat in which the cysteine in position 22 was replaced by a serine (Tat Oyi) making its transactivation silent
[84]. Similarly, in the cART era, progressive accumulation over time of proviruses defective for replication
[85] and/or for gene expression (i.e., containing solo LTRs)
[86] was found. This is probably the result of the combined pressure exerted by cART and by a sufficiently restored immune response against HIV. In this scenario, only silent proviruses lacking Tat, as solo LTRs, are destined to persist.
4.3. Establishment and Maintenance of Latent Virus Reservoirs
Despite cART effectiveness, a low-level intermittent residual plasma viremia (<50 copies per mL), as well as viral “blips” (50–1000 copies/mL), persist, predict virus rebound and are believed to be at the origin of persistent immune activation, residual disease and the onset of comorbidities in treated patients (reviewed in
[13]). In fact, HIV gene expression and viral production are sporadically resumed in HIV latently infected cells constituting the HIV reservoirs
[3][4][3,4].
Furthermore, HIV gene expression it is not halted by cART, whereas residual virus replication, driven by low drug penetration in lymphoid tissue compartments
[87][88][87,88] and drug-resistant cell-to-cell transmission modalities
[89], may occur
[90]. Of importance, while Tat mRNA accumulates in the nuclei of resting CD4 T cells from peripheral blood and cannot support HIV protein expression
[91], resting CD4 T cells from lymphoid tissues do express positive transcription elongation factor (PTEF)b1 and support HIV-1 gene expression and replication, allowing HIV acquisition at the portal of entry
[92] and likely contributing to residual virus replication and reservoir replenishment.
Intracellular HIV-1 Tat plays a key role in virus reservoir establishment and maintenance. In fact, Tat expression enhances stochastic fluctuations of the basal HIV transcriptional machinery driven by host transcriptional factors
[54][93][94][54,93,94]. This, in turn, drives Tat expression itself into stochastic oscillations around the threshold of virus transcriptional extinction. Consequently, the Tat positive transcriptional feedback loop is pivotal for fate decision-making between HIV productive and latent infection
[54][93][94][54,93,94]. The stochastic features of the Tat circuitry may in part explain the failure of “shock and kill” strategies to eradicate HIV based on the deterministic reversion of HIV latency through latency-reversing agents
[95]. In contrast, agents inhibiting the Tat transcriptional loop effectively block the reactivation of latent HIV, which has led to the “block and lock” strategy for a permanent shut-off of HIV reservoirs
[96].
Not surprisingly, Tat is produced and released in treated patients
[52][97][98][52,97,98], pointing to a role for eTat in HIV reservoir dynamics. As already mentioned, eTat crosses the blood–brain barrier and chemoattracts monocytes/macrophages and T cells, promoting inflammation, the permissivity of resting T cells and T-cell activation,
[52]. Thus, eTat promotes the establishment and maintenance of an HIV reservoir in the CNS, which is relatively insensitive to immune control and ART
[53]. In this context, several lines of evidence also suggest that eTat plays a key role in the establishment and maintenance of the memory CD4 T cell reservoir. In fact, eTat promotes the activation and differentiation of naïve CD4 T cells towards the effector-memory phenotype
[99], thus increasing the frequency of cells transitioning from the activated to the resting state, which, in turn, is associated with HIV latent infection
[100]. Further, it upregulates anti-apoptotic genes, particularly Bcl-2, in CD4 T cells, promoting their survival
[101]. Taken together, it is apparent that eTat is pivotal not only in HIV acquisition and spreading, but also in the establishment and maintenance of reservoirs.
Extensive evidence of the role of intracellular Tat in HIV reservoirs has been reviewed elsewhere
[102].