Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Teodor Ioan Bajeu and Version 2 by Mona Zou.
Restenosis refers to the narrowing of a blood vessel’s diameter following an angioplasty procedure. Intrastent restenosis (ISR) is a challenging medical problem. A meta-analysis showed that percutaneous coronary intervention (PCI) for ISR is associated with a higher incidence of adverse cardiac events than PCI for de novo lesions. This happens especially because of a higher incidence of risk-adjusted major adverse cardiac events compared with PCI for de novo lesions at a median of ≈30 months.
- intrastent restenosis
- bare-metal stents
- drug-eluting stents
- pathophysiology
- intravascular imaging
- risk factors
- coronary artery disease
- treatment
- drug-eluting ballons
- angiography
1. ISR—Definition, Incidence, and Pathophysiology
ISR is characterized by a progressive luminal constriction within the stent, predominantly manifesting within the timeframe of 3 to 12 months subsequent to stent angioplasty [1][9]. From a clinical perspective, this phenomenon manifests as recurrent angina or heart failure [2][10]. It is noteworthy that it may also manifest as acute myocardial infarction in approximately 10% of afflicted patients [3][7]. In contrast, intrastent thrombosis constitutes an acute thrombotic occlusion, representing a dire event with the potential for precipitating sudden cardiac death or protracted acute myocardial infarction [4][11]. Despite the early revascularization, the mortality at 6 months is very high in this case [1][9].
The incidence of ISR is contingent upon various factors, including the stent type, the intricacy of the stented lesions, and the presence of risk factors [5]. Significantly elevated restenosis rates are observed in instances characterized by lesion complexity, such as those involving small vessels, extended lesions, or bifurcation lesions [6][12]. Determining the incidence of ISR is challenging due to its dependence on a multifactorial and variable interplay of factors. During the period in which balloon angioplasties were used, restenosis rates ranged from 32% to 55% among all procedures, subsequently declining to a range of 17% to 41% [7][8][9][10][13,14,15,16] with the advent of bare-metal stents [11][12][13][17,18,19]. A pivotal achievement in addressing this complication was the adoption of DESs, leading to a reduction in restenosis rates to levels below 10% [14][15][20,21]. ISR seems to manifest more frequently in patients suffering from multivessel disease compared to those affected solely by single-vessel disease [16][22].
ISR refers to a condition characterized by a significant reduction, typically equal to or exceeding 50%, in the diameter of the coronary lumen [17][23].
The advent of coronary stents has substantively transformed the therapeutic landscape for individuals afflicted by both chronic coronary syndromes and acute coronary syndromes [18][19][24,25]. Nevertheless, the widespread adoption of percutaneous revascularization procedures has ushered in a novel pathological phenomenon: ISR. The pathophysiology of ISR is a complex process involving many cellular mechanisms and depends on a series of risk factors [20][26].
It is characterized by the re-narrowing of a coronary artery at the site of a previously implanted stent [21][27]. The predominant mechanism underlying ISR is characterized by tissue proliferation, commonly referred to as neointimal hyperplasia [22][28]. This process reflects an exaggerated homeostatic healing response triggered by the arterial wall injury incurred during stent implantation, as the literature delineates [23][29]. The causative factors implicated in this phenomenon encompass localized inflammation arising from mechanical injury to the intimal and medial layers. This subsequently drives an aggressive neointimal hyperplasia marked by the proliferation of smooth muscle cells and the deposition of extracellular matrix [24][25][30,31]. Moreover, hypersensitivity reactions to the metallic components and polymer constituents intrinsic to early-generation DESs have been established as recognized mechanisms contributing to neointimal hyperplasia [26][32].
Coronary atherosclerotic disease is most commonly managed through the intervention of angioplasty with stent placement, a therapeutic modality that, while lifesaving, has been associated with adverse events that curtail its enduring efficacy [27][33]. Technical inadequacies, exemplified by factors such as stent malposition or inadequate expansion, resulting in the establishment of laminar blood flow patterns, subsequently promote the development of neointimal hyperplasia [28][34]. After angioplasty with a stent, a prevailing concern in long-term progression is the occurrence of intrastent neo-atherosclerosis [29][35]. Notably, the utilization of old-generation bare-metal stents has been constrained in adherence to contemporary medical practice guidelines due to a notable incidence of ISR, which has historically ranged from 17% to 41% [2][10]. While the introduction of new-generation DESs has substantially reduced the rate of such complications [14][20], DES-related failures persist as a matter of concern, affecting up to 10% of all implanted stents [14][15][20,21].
As mentioned before, using BMSs leads to a high rate of restenosis that can reach 40%. The principal etiology of this complication is commonly attributed to the incremental expansion of the extracellular fibrotic matrix, resulting in a gradual reduction in the luminal diameter [14][20]. In the contemporary era marked by the utilization of DESs, the pathogenesis of ISR appears to be contingent upon several interrelated factors, including the host’s heightened material sensitivity, the suppression of healing processes due to antiproliferative medications, and the organism’s reaction to stent implantation [30][31][36,37] (Figure 1). The pathophysiology of ISR (Figure 2) appears to involve a complex cascade of events, including the hyperplasia of smooth muscle cells, the migration of pro-inflammatory cells, the recruitment of marrow progenitors, including bone-marrow-derived progenitor cells (BMPCs), and the proliferation of the extracellular matrix, as suggested by the existing literature [30][31][36,37]. Hence, the interplay between the systemic pro-inflammatory response and the concurrent local pro-inflammatory response induced by the endothelial disruption during stent implantation collectively orchestrates the vascular healing process [32][38]. This dynamic interaction culminates in forming a neointimal zone characterized by the proliferation of unregulated smooth muscle fibers, ultimately contributing to the clinical manifestation of restenosis [33][39]. At the local level, the injury induced by stent deployment initiates a multifaceted cascade of processes, commencing with endothelial disruption and subsequent exposure of the intimal layer, which imparts a prothrombotic effect [32][38]. This local inflammatory milieu subsequently triggers the release of cytokines and growth factors, activates platelets, and fosters the proliferation and migration of smooth muscle fibers. These alterations can culminate in two potential outcomes: vascular healing or pathological progression. The latter pathological process, in particular, may eventually predispose individuals to ISR. Endothelial activation, prompted by cellular injury induced by the mechanical impact of the stent, initiates a cascade of events, including platelet activation. Notably, platelets are often the primary cells to respond to stent placement [34][35][40,41].
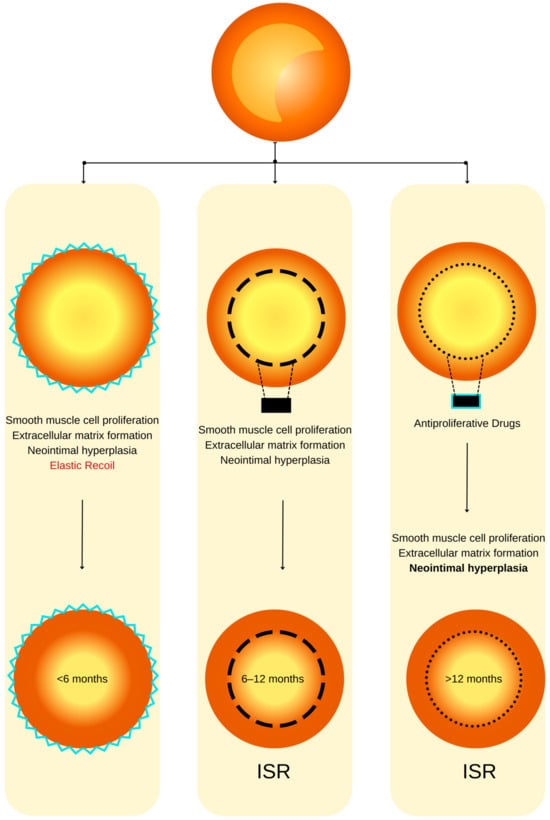
Figure 1. ISR physiopathology (adapted from an open–access source [26][32]). (Left): Coronary restenosis after conventional balloon angioplasty primarily arises from the phenomenon of elastic recoil of the arterial vessel wall. Moreover, the trauma inflicted on the coronary artery triggers the initiation of smooth muscle cell proliferation, their migration, and the deposition of an extracellular matrix. This cascade of events culminates in the formation of neointimal hyperplasia, which ultimately contributes to the pathogenesis of restenosis. (Center): The deployment of a BMS is associated with a heightened level of vascular injury, thereby augmenting the magnitude of neointimal hyperplasia and elevating the risk of in-stent restenosis. (Right): DESs dispense antiproliferative agents, effectively mitigating the extent of neointimal hyperplasia and correspondingly diminishing the susceptibility to in-stent restenosis.
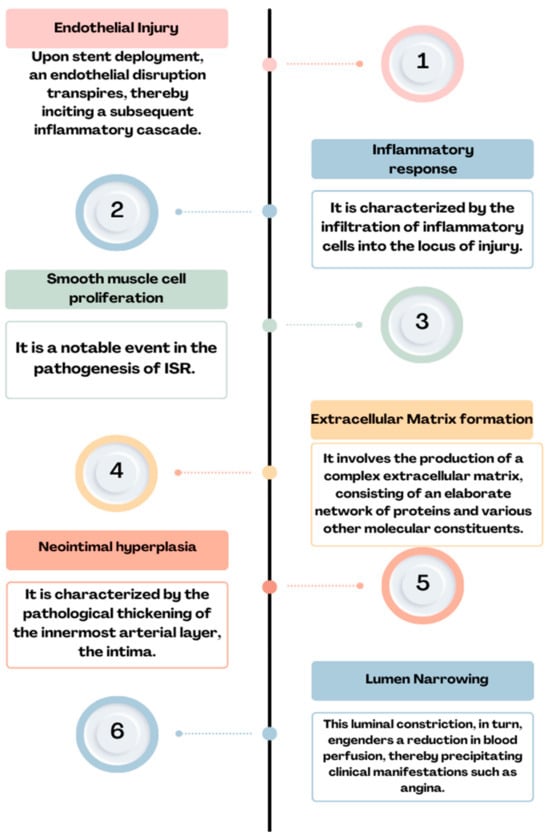
Figure 2. Physiopathology of ISR: endothelial injury→ inflammatory response→ smooth muscle cell proliferation→ extracellular matrix formation→ neointimal hyperplasia→ lumen narrowing = ISR.
After arterial injury, platelets promptly adhere to the affected site and initiate the release of thromboxane A2. The glycoprotein (GP) IIb/IIIa complex engages with fibrinogen, facilitating platelet aggregation and activation [36][42]. Furthermore, activated platelets release many bioactive factors, among which the platelet-derived growth factor (PDGF) is prominent. PDGF exhibits both mitogenic and chemotactic properties, significantly influencing smooth muscle cells [37][43]. It contributes to the induction of oxidative stress, thereby facilitating the transition of smooth muscle cells from a contractile phenotype to a synthetic one [38][44]. The release of histamine from mast cells and platelets has been implicated in the pathogenesis of intimal hyperplasia [38][44]. This assertion supports studies involving porcine models of endothelial dysfunction, wherein a substantial 20- to 90-fold elevation in histamine concentration has been documented [39][45]. Interleukin-1, when released by platelets, triggers an upregulation in the production of interleukin-6 and interleukin-8, exerting pro-inflammatory effects. This cascade of events includes the stimulation of smooth muscle fiber migration, achieved through the activation of actin polymerization and the initiation of tyrosine phosphorylation at the cytoskeletal protein level, particularly in association with focal adhesion and smooth muscle fiber proliferation [32][38]. Extracellular vesicles emanating from platelets elicit a response in smooth muscle fibers, inducing the production of interleukin-6 while concurrently triggering the expression of αIIbβ3 and P-selectin. These cellular changes promote interactions between smooth muscle fibers and monocytes [40][46].
Furthermore, platelet-derived microvesicles have been demonstrated to induce endothelial protein C receptor (EPCR) proliferation characterized by the presence of von Willebrand factor (vWF+) and CD34+ markers. This effect is mediated through the release of transforming growth factor (TGF)-β1, as evidenced in a rat arterial injury model [41][47].
Within the vessel wall, the coexistence of an inflammatory response stemming from the presence of the foreign body, coupled with the anti-inflammatory effect exerted by the drug released from the stent, collectively engenders a deceleration in the process of reendothelialization. The exposure of adhesion molecules, such as P-selectin, serves as a stimulus for the recruitment of corresponding monocytes and the subsequent secretion of pro-inflammatory cytokines, namely interleukins 6 and 8. This cascade of events culminates in the infiltration of neutrophils, monocytes, and macrophages into the subendothelial space [32][38]. Elevated levels of monocytes and eosinophils at the three-month mark following PCI serve as predictive factors for late ISR after deploying a pharmacologically active stent [42][48]. While the precise involvement of mast cells in the pathophysiology of restenosis remains a subject of ongoing investigation, it is noteworthy that mast cells have been observed to release chymase [43][49], an enzyme implicated in generating angiotensin II and tumor growth factor-β. This consequential cascade of molecular events subsequently fosters fibroblast proliferation and is associated with the development of neointimal formation [44][45][50,51]. Experimental evidence in animal models has demonstrated the efficacy of chymase expression inhibitors in attenuating neointimal formation [46][52].
Neo-atherosclerosis represents a mechanistic phenomenon observed in the context of DES utilization, and its incidence is on the rise. This process is typified by the accretion of lipid-laden foamy macrophages, occasionally accompanied by a necrotic core predominantly localized within the stent deployment region [29][35].
2. Risk Factors for ISR
2.1. Patient-Related Factors
The clinical prognosticators associated with in-stent restenosis (ISR) encompass a spectrum of factors, comprising, but not limited to, chronic kidney disease, elderly age, male gender, diabetes mellitus, and elevated body mass index, among various others [47][53]. Furthermore, established cardiovascular risk factors associated with atherosclerosis, such as hypertension, smoking, and dyslipidemia, have been evidenced to exert a pathogenetic influence on the development of neointimal hyperplasia [48][54].
2.2. Clinical Factors
ISR is a multifactorial phenomenon, subject to variability contingent upon individual patient characteristics, angiographic predictors, and the intricacies associated with the angioplasty procedure [49][55]. Notably, in the context of diabetic patients, it is imperative to underscore that the second generation of DESs exhibit a notable susceptibility to ISR, manifesting with an incidence rate of 8.7% [50][56]. In contrast, non-diabetic patients present a relatively diminished propensity for ISR, as evidenced by a prevalence of 5.7% [29][35].
Concerning gender-based prevalence, a study published in 2023 employed intracoronary imaging, specifically, optical coherence tomography (OCT), to demonstrate that the incidence of ISR exhibits a greater frequency among male patients [51][57]. Hence, the imaging data reveal a notable discrepancy, whereby men exhibit a significantly elevated risk in contrast to women, characterized by a higher incidence of thin-cap fibrous atherosclerosis (TCFA) (37.4% [n = 77] vs. 9.3% [n = 4], p < 0.001), as well as in-stent neo-atherosclerosis (ISNA) (82.0% [n = 169] vs. 62.8% [n = 27], p = 0.005).
Another study, conducted by Bajdechi et al. and published in 2023, explored a distinct patient cohort, specifically, individuals afflicted with Human Immunodeficiency Virus (HIV), revealing their heightened susceptibility to acute coronary events. This elevated risk can be attributed, in part, to the influence of antiviral medications and the concurrent discordant inflammatory response [52][58]. While patients afflicted with HIV exhibit an elevated risk of recurrent acute coronary syndrome, it is noteworthy that the stage of the disease does not exert a discernible influence on the prevalence of ISR [52][58].
Arterial hypertension, identified as an independent cardiovascular risk factor [53][59], was shown for the first time to be responsible for intrastent restenosis in a retrospective study involving 796 patients who underwent angiography due to angina recurrence or reversible myocardial ischemia [54][60]. The study revealed that effective blood pressure control during the initial PCI procedure correlated with a 24% lower risk of ISR. Additionally, factors such as total cholesterol levels, the use of beta-blockers or antiplatelet agents, and the site of stent implantation were linked to further reductions in ISR, particularly among patients with BMSs [54][60].
Moreover, it is well established that the cessation of antiplatelet medications ranks among the primary risk factors associated with intrastent restenosis. Consequently, non-adherence to pharmacological treatment regimens represents an additional significant risk factor for the development of ISR [55][61]. Additionally, individuals presenting with hypertension and a diagnosis of heart failure manifest an elevated susceptibility to ISR [56][62].
Furthermore, in a retrospective observational study, the incidence of restenosis in the coronary stent group was higher compared to the non-stent group when considering factors such as a family history of CHD, a history of type 2 diabetes, hypertension, smoking, drinking, withdrawal of aspirin, use of conventional doses of statins, calcified lesions, having ≥3 implanted stents, stent length ≥30 mm, and stent diameter <3 mm [57][63].
2.3. Angiographic Factors
As evidenced by the existing literature, the deployment of a stent in a vessel elicits a localized inflammatory response. This inflammatory cascade constitutes a pivotal component in the genesis of a pathological phenomenon recognized as ISR [58][64]. Furthermore, the intricacies of this multifaceted phenomenon extend beyond the mere presence of a stent, encompassing an array of stent-dependent, intra-stent, and extra-stent risk factors. The schematic delineation portrayed in Figure 3 illustrates the angiographic patterns under discussion. It is imperative to underscore that this classification holds pivotal significance in prognosis and expeditious patient triage for both clinical and investigative objectives [59][65].
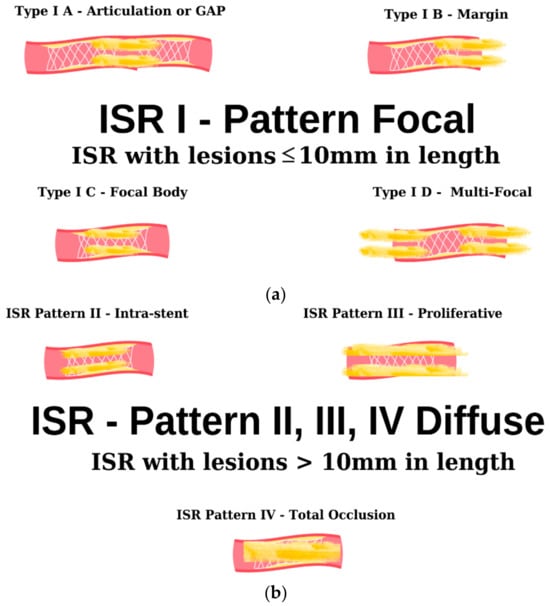
Figure 3. Angiographic classification of intrastent restenosis (adapted from an open-access source [59][65]). (a) Type I: there are four types of focal ISR described in the image above. (b) Type II: the observed lesions are confined strictly to the confines of the stent and do not exhibit any extension beyond its proximal or distal extremities. Type III: Diffuse, proliferative ISR. The identified lesions manifest a length exceeding 10 mm, exhibiting an extension that surpasses the boundaries of the stent at both its proximal and distal margins. Type IV: total occlusion of the stent resulting in no coronary perfusion.
In a retrospective study [60][66], the utilization of BMSs was associated with an ISR rate of 30%. Subsequently, with the advent of second-generation DESs, this proportion exhibited a notable reduction, declining to 12%. Consequently, the use of BMSs has been identified as a risk factor for ISR, thereby underscoring the rationale behind its diminishing prevalence in contemporary clinical practice in favor of the more favored DESs.