Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ahmed M. Agiba and Version 2 by Camila Xu.
Light-triggered mechanisms that can be exploited to release encapsulated drugs from liposomes are photoisomerization, photocleavage (photo-oxidation), surface plasmon resonance absorption (photothermal activation), photochemical hydrophobicity change (photochemical activation), and photo-crosslinking and de-crosslinking.
- smart nanocarriers
- light-responsive liposomes
- light-triggering mechanisms
1. Introduction
1.1. Liposomes as Drug Nanocarriers
Nanocarriers were first discovered in the early 1960s, when scientists proposed the application of liposomes for drug delivery [1]. Since then, many nanocarrier systems have been developed and approved for marketing by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA). Nanocarriers have been extensively used in drug delivery owing to their exceptional physicochemical properties, such as nanometric particle size, surface charge, high entrapment efficiency, and drug loading capacity [2]. Nanocarriers used in cancer treatment have received particular attention from researchers worldwide, since most conventional chemotherapeutic drugs cause systemic toxicity resulting from their poor stability in biological systems, non-selectivity, and non-specificity toward cells expressing the targeted receptors [3]. The first evidence on the feasibility and effective use of nanocarriers in cancer treatment was reported in 1976 by Langer et al. [4], who prepared the first sustained-release, long-circulating poly(ethylene glycol)-poly(lactic acid-ethanolic acid) (PEG-PLEA) nanoparticles, which were later approved by the FDA as the first nanomedicine for therapeutic use in cancer treatment. Generally, nanocarriers are categorized into two main classes: organic and inorganic nanocarriers [5][6][5,6]. Organic nanocarriers include nanoemulsions (10–1000 nm), nanosuspensions (<1 µm), nanoliposomes (50–450 nm), polymeric nanoparticles (10 nm to 1 µm), solid-lipid nanoparticles (10–1000 nm), and nanodendrimers (15–200 nm), while inorganic nanocarriers include gold nanoparticles (5–400 nm), silver nanoparticles (1–100 nm), mesoporous silica nanoparticles (30–300 nm), and superparamagnetic iron oxide nanoparticles (100 nm to 5 µm). These tumor-targeting, nano-sized drug delivery systems were developed primarily to reduce the systemic toxicity of chemotherapeutic drugs through encapsulation into nanocarrier systems, which allowed for site-specific delivery with improved passive and active drug targeting (i.e., disease-specific targeted therapeutics). Of all these nanocarriers, liposomes are very promising drug delivery systems with the advantages of being non-toxic, biocompatible, and biodegradable [2]. Liposomes were first discovered by Bangham et al. [7] in 1964. They discovered how membrane molecules interact with water to form unique structural forms, which were described as swollen phospholipid systems [7]. Briefly, liposomes are defined as vesicular systems consisting of one or more concentric spheres of phospholipid bilayers separated by aqueous or buffer compartments [7][8][7,8]. When phospholipids are dispersed in an aqueous medium like water or buffer, the hydration of phospholipid polar heads results in a heterogeneous mixture of spherical structures, generally referred to as vesicles, most of which contain multiple phospholipid bilayers forming concentric spherical shells [7][8][7,8]. Those were the liposomes first reported by Bangham et al. [1][7][8][1,7,8], nowadays referred to as multilamellar large vesicles (MLVs). The sonication of these lipid dispersions results in the size reduction of these liposomes to vesicles containing only a single bilayer with diameters ranging from 20 to 100 nm, later referred to as small unilamellar vesicles (SUVs) [9]. Large unilamellar vesicles (LUVs, 100–1000 nm) are intermediate in size between MLVs (>700 nm) and SUVs [9]. The main components of liposomes are phospholipids and cholesterol, which are naturally occurring substances [9][10][9,10]. Phospholipids are amphiphilic molecules with hydrophobic non-polar tails and hydrophilic polar heads. These amphiphilic molecules spontaneously organize into liposomes in an aqueous or buffer environment, driven by hydrophobic interactions and other intermolecular interactions [11]. The proper choice of phospholipid is important to achieve the desired effects. Table 1 shows the most commonly used phospholipids in the preparation of liposomes (data extracted from the Sigma-Aldrich (Burlington, MA, USA) and Avanti Polar Lipids (Alabaster, AL, USA) databases). Figure 1 shows the classification of liposomes according to their structures, sizes, compositions, and preparation methods.
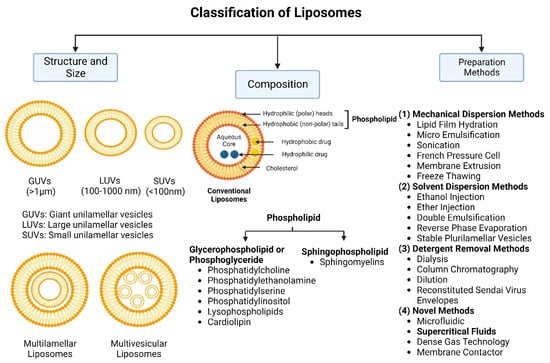
Figure 1.
Classification of liposomes according to their structures, sizes, compositions, and preparation methods.
Table 1. Lipids used for the preparation of liposomes (data extracted from the Sigma-Aldrich Aldrich (Burlington, MA, USA) and Avanti Polar Lipids Alabaster, AL, USA) databases).
| Lipid Name and CAS No. | Synonym | Molecular Formula | Chemical Structure |
|---|---|---|---|
| Neutral | |||
| Cholesterol (CAS No.: 57-88-5) |
--- | C27H46O | 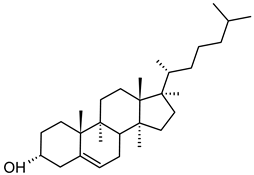 |
| Anionic | |||
| 1,2-Dimyristoyl-sn-glycero-3-phosphoglycerol, sodium salt (CAS No.: 200880-40-6) |
DMPG-Na | C34H66NaO10PNa | 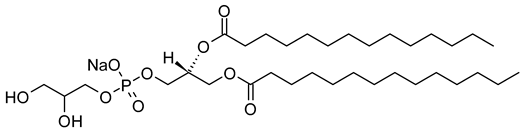 |
| 1,2-Dipalmitoyl-sn-glycero-3-phosphoglycerol, sodium salt (CAS No.: 200880-41-7) |
DPPG-Na | C38H74NaO10PNa | 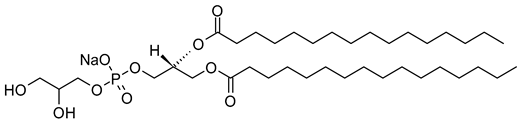 |
| 1,2-Distearoyl-sn-glycero-3-phosphatidylglycerol, sodium salt (CAS No.: 200880-42-8) |
DSPG-Na | C42H82NaO10PNa | 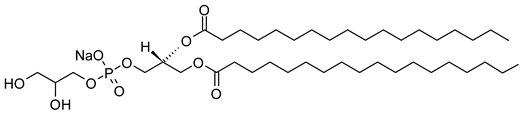 |
| N-(Methoxypolyethylene glycol 5000 carbamoyl)-1,2-dipalmitoyl-sn-glycero-3-phosphatidylethanolamine, monosodium salt (CAS No.: 205494-72-0) |
MPEG-5000-DPPE-Na | ||
Figure 2.
Supercritical CO
2
(SC-CO
2
)-assisted liposome formation.
Numerous liposomal formulations have been clinically approved for human use to treat cancer and other chronic diseases, and are currently available on the global pharmaceutical market. Table 2 shows FDA- and EMA-approved liposomal drug formulations [20][21][20,21]. The Orange Book identifies drug products approved on the basis of safety and effectiveness by the FDA [20]. The electronic medicines compendium (EMC) identifies drug products approved for human use in the UK and Europe [21].
| Product Name | Approval Date | Product Description | Liposome Composition | Indication and Usage | Manufacturer |
|---|---|---|---|---|---|
| Doxil® | FDA: 1995 EMA: 1996 |
Doxorubicin encapsulated in stealth liposomes. | MPEG-DSPE, HSPC, cholesterol. | Ovarian cancer, AIDS-related Kaposi’s sarcoma. | Janssen Pharmaceuticals (Beerse, Belgium) |
| (C | |||||
| 2 |
Figure 3) [22]. Since targeting the overexpressed surface receptors of cancer cells in order to enhance cellular uptake and intracellular activity is a promising approach, several cell surface strategies have been developed so far, aiming towards achieving the targeted inhibition of these receptors [22][24][22,24]. The efficiency of active targeting and ligand receptor interaction is dependent on certain factors [25], such as (1) the extent of receptor expression level on tumor cells relative to non-tumor cells, (2) the availability of surface receptors on tumor cells, (3) the internalization rate, and (4) the heterogeneity of receptor expression in tumor cells. Active targeting can be achieved through the surface engineering of liposomes via decoration with aptamers (oligonucleotides), carbohydrates, glycoproteins, monoclonal antibodies (mAbs) and their fragments, peptides, proteins, or other small molecules adsorbed onto the liposomal surface [22][24][22,24]. Figure 3 shows the surface modification of liposomes for active targeting. Figure 4 shows the distinction between passive and active targeting. Table 3 shows some examples of liposomes and their ligands used for active targeting.
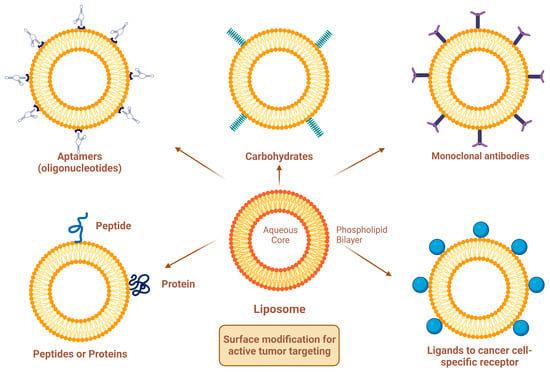
Figure 3.
Surface modification of liposomes for active targeting.
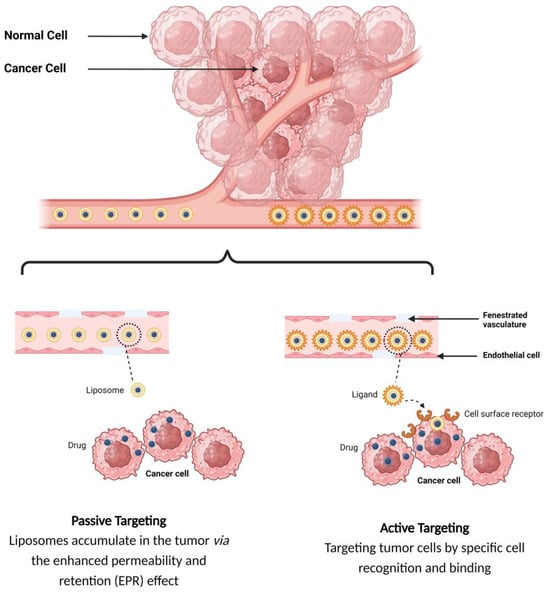
Figure 4.
Targeting mechanisms of liposomes.
Table 3.
Examples of liposomes and their ligands used for active targeting.
| Active Targeting Ligand | Encapsulated Drug | Preparation Method | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Abelcet® | FDA: 2005 | Amphotericin B lipid complex injection. | DMPC, DMPG. | Invasive fungal infections. | Leadiant Biosciences, Inc. (Gaithersburg, MD, USA) | |||||
| DaunoXome® | ||||||||||
| PEGylated Liposomes | ||||||||||
| mAbs (MM-302) | Doxorubicin | Thin-film hydration | [26] | |||||||
| FDA: 1996 | EMA: 2004 |
Daunorubicin encapsulated in liposomes. | DSPC, cholesterol. | Advanced HIV-associated Kaposi’s sarcoma. | Galen Ltd. (Craigavon, UK) | |||||
| mAbs (Sortagged anti-EGFR) | Doxorubicin | Ethanol injection | [27] | AmBisome® | FDA: 1997 EMA: 2006 |
Amphotericin B liposome for injection. | HSPC, cholesterol, DSPG, alpha tocopherol. | Cryptococcal meningitis in HIV-infected patients. | Gilead Sciences, Inc. (Foster City, CA, USA) | |
| H | ||||||||||
| 4 | ||||||||||
| Folate | Oleuropein | Thin-film hydration | [28] | DepoCyt® | FDA: 1999 EMA: 2001 |
Cytarabine liposome injection. | Cholesterol, triolein, DOPC, DPPG. | Lymphomatous meningitis. | Pacira Pharmaceuticals, Inc. (Parsippany, NJ, USA) | |
| Folate | Rapamycin | Thin-film hydration | [29] | Myocet® | FDA: 2000 EMA: 2000 |
Non-PEGylated liposomal doxorubicin. | Phosphatidylcholine, cholesterol. | Metastatic breast cancer in adult women. | Teva Pharmaceuticals (Tel Aviv, Israel) | O)n |
| Folate | Arsenic trioxide | Thin-film hydration | [30] | Mepact® | FDA: 2001 EMA: 2009C39H76NO10PNa |
A liposomal suspension of mifamurtide.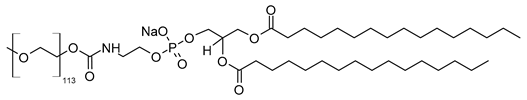 |
||||
| POPC, OOPS. | High-grade resectable non-metastatic osteosarcoma. | Takeda Pharmaceutical Company (Tokyo, Japan) | ||||||||
| Transferrin | Plumbagin | Thin-film hydration | [31] | N-(Methoxypolyethylene glycol 2000 carbamoyl)-1,2-dipalmitoyl-sn-glycero-3-phosphatidylethanolamine, monosodium salt (CAS No.: 384835-61-4) | ||||||
. However, despite all these advantages, PEG-functionalized liposomes showed some serious drawbacks. For instance, PEG demonstrates potential immunogenicity owing to the activation of complement in response to antibodies [42][43][42,43]. In fact, PEG-based delivery systems support the phenomenon of accelerated blood clearance (ABC), owing to the formation of anti-PEG immunoglobulin M (IgM) antibodies by the spleen after initial administration. Following the second administration, the anti-PEG IgM binds to PEG groups on the surface of liposomes, resulting in the activation of the complement system, which subsequently leads to opsonization of the liposomes by C3 fragments and consequently enhances the cellular uptake of liposomes by the Kupffer cells in the liver, which in turn greatly affects the drug’s bioavailability [43]. Even though the ABC phenomenon poses a significant challenge for certain drugs, it does not pose a critical problem for PEGylated liposomes used in cancer therapy, owing to the high lipid content of liposomes encapsulating anticancer cytotoxic agents [43].
1.3.2. Ligand-Functionalized Liposomes
The adsorption of active targeting moieties on the surface of liposomes has played a significant role in enhancing liposomal accumulation in cancer cells since this structural characteristic helps to increase the therapeutic index of the encapsulated drug, maximize on-target effects, and minimize off-target effects. Briefly, active targeting is a surface modification process where active targeting moieties are adsorbed onto the liposomal surfaces, which substantially help to recognize and bind specifically to target cells through ligand–receptor interactions [44]. Actively targeting liposomes are made by grafting moieties such as aptamers, carbohydrates, glycoproteins, mAbs and their fragments, peptides, proteins, and small molecules adsorbed onto the liposomal surfaces (Figure 3). The targeting moiety can be either inserted directly into the lipid membrane or attached specifically to the distal end of the polymer [38][45][38,45]. Aptamers are short single-stranded oligonucleotides of deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) with typically 20–100 base pairs. They have been widely studied as targeting agents to deliver various drugs and photosensitizers with high specificity to cell receptors and binding affinity at the nanomolar level [46]. Aptamers can be easily synthesized chemically and modified for conjugation. Different aptamers recognize different molecular targets. Cell-type-specific aptamers can be designed and synthesized by the exponential enrichment (SELEX) method, which can generate cell-type-specific aptamers with a high target affinity [47]. Transferrin is a blood plasma glycoprotein that plays a key role in iron metabolism by binding iron and transporting it into cells via TFR-mediated endocytosis [48]. Tumor cells often require excess amounts of iron for fast proliferation; thus, transferrin receptors on tumor cells are overexpressed in order to increase the uptake of iron from the plasma [48][49][48,49]. Folate (folic acid) is an essential vitamin for DNA synthesis and cell division. Folate and its derivatives have a high affinity for FRs and are internalized by FR-mediated endocytosis [50]. The receptor–ligand interaction affects the rate of cellular internalization, which in turn influences the accumulation of liposomal drugs in the tumor cells. Folate has the advantages of fast internalization and intracellular recycling rate, which subsequently fastens the delivery of drugs and photosensitizers to tumor cells [50][51][50,51]. Antibodies can recognize tumor cells by binding to specific antigens overexpressed by tumor cells with high affinity. Antibody targeting has been extensively studied for the delivery of drugs and photosensitizers to tumor cells; however, the therapeutic uses of mAbs are limited owing to antibody recognition and immunologic response [52]. Antibody fragments, such as single-chain variable fragments (scFv) and antigen-binding fragments (Fab), have been developed to modulate the overexpression of specific membrane antigens. Antibody fragments are smaller in size than mAbs, which allows them to efficiently reach their targets [52]. Although peptides have low molecular weights and a lower immunogenicity than antibodies, which lead to improved tumor penetration, they have a lower binding affinity. Peptides can be easily synthesized chemically and modified to obtain the desired physicochemical and biopharmaceutical properties. However, the presence of PEG polymers on the liposome surface may prevent peptides from interacting with cells. Therefore, peptides should be projected away from the liposome surface to avoid the shielding of the PEG polymer [53]. To conclude, the proper selection of targeting ligands is necessary to achieve high binding affinity and specificity. A recent trend in liposome surface functionalization includes the decoration of the liposomal surface with two ligands (i.e., dual-targeting). Dual-targeted liposomes offer numerous advantages, such as targeting multiple receptors, delivering more than one drug to target sites, enabling the encapsulated drugs to exert enhanced therapeutic effects, and reducing normal tissue toxicity and damage [54][55][54,55]. Two strategies are commonly used for dual-targeting based on different ligand combinations. The first strategy is based on the combination of two targeting ligands that leads to improved tumor selectivity and the cellular uptake of liposomes by specific tumor cells. The second strategy is based on the combination of targeting ligands with cell-penetrating peptides (CPPs) to achieve selective cellular uptake and enhanced anticancer efficacy toward specific tumor cells [55][56][55,56]. A dual-ligand combination involves the use of two ligands to target different receptors, which can be expressed only on one cell or on different cells. These ligands can be combined into one molecule or even placed separately on the surface of liposomes [56]. Common examples of dual-ligand combinations are those in which the second ligand is combined with either RGD, HA, or transferrin, since those ligands are overexpressed on different tumor cells [56]. In general, dual-ligand combinations can be classified into three different classes based on the types of target cells and the target sites of action. In the first class, two ligands are used to target one cell type, which is overexpressed on two receptors. The second class involves the use of two ligands to target two cell types, while the third class combines cell membrane targeting with intracellular organelle targeting (such as nuclear targeting or mitochondrial targeting) [55][56][55,56]. In fact, surface-modified liposomes with one or more targeting ligands exhibit improved cellular uptake compared with conventional liposomes due to receptor-mediated endocytosis. However, the cellular uptake efficiency of liposomes by tumor cells is still limited since the receptors on the surface of tumor cells dynamically change with tumor progression, leading to the saturation of receptor–ligand binding [56][57][56,57]. The discovery of CPPs helped to overcome these barriers, especially the cell membrane barrier, and subsequently enhanced cellular uptake [57][58][57,58]. However, in vivo applications of surface-functionalized liposomes with CCPs are limited since the non-specificity of CPPs may penetrate normal cells and lead to systemic toxicity [59][60][59,60]. Furthermore, the surface-functionalized liposomes with CCPs may be easily recognized by the RES owing to the high density of surface positive charge [61].
1.3.3. Stimuli-Responsive Liposomes
Liposomes can respond to different internal and external stimuli and thus trigger the release of encapsulated drugs in a controlled manner to specifically target cancer cells. Internal stimuli include enzymes (such as cathepsin B enzyme, a lysosomal protease of the papain family, which is overexpressed in several pathological conditions and malignancies, including the brain, breast, colon, lung, and prostate cancers), pH (such as DOPE that adopts an inverted hexagonal phase II (HII phase) at low pH and a bilayer structure (Lα phase) at neutral pH to promote the liposomal membrane permeabilization), redox (e.g., disulfide-containing liposomes that can be easily broken down by reducing glutathione at the tumor site, leading to the complete disintegration of the liposomal membrane), and temperature (i.e., temperature can act either as an external stimulus when heat source is applied from outside, or can be internal when cancer pathological lesions have a naturally elevated temperature) [62][63][62,63], while external stimuli include light, electrical fields, magnetic fields, and ultrasound waves [62][63][62,63]. Light, in the UV-visible-IR region, is a very promising tool for biological and medical applications due to its non-invasive nature, high spatial resolution and temporal control, tuneability over a wide range of wavelengths, convenience and ease of application, and robustness [64]. In comparison with other stimuli, light provides an unparalleled spatiotemporal modulation of molecular processes [65], making it highly suitable for clinical and therapeutic applications [64][65][64,65]. Table 4 summarizes the advantages and limitations of different types of stimuli. Light-responsive liposomes have been recently introduced as smart, intelligent drug targeting delivery systems to target drugs for specific sites with high spatial and temporal control over drug release [66]. These systems utilize nonionizing radiation and are mainly composed of biocompatible, biodegradable materials that can be straightforwardly tailored to the target sites for clinical and therapeutic applications [66]. Although most light-responsive liposomes respond to UV irradiation that has a poor tissue penetration and high phototoxicity, optical technologies like laparoscopic tools are now commonly used for reaching deeper located tissues [64][65][64,65]. On the other hand, NIR is safer for use, causes less cell damage, and has good tissue penetration. However, the lower energy of NIR may not be efficient enough to induce the desired drug release response from liposomes [67][68][67,68].
Table 4.
Comparison between different types of stimuli.
| Stimuli | Advantages | Limitations | References | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Light |
|
|
[64][65][66][67][68] | ||||||
| Heat |
|
|
[69][70] | ||||||
| pH |
|
|
[71][72] | ||||||
| MPEG-2000-DPPE-Na | |||||||||
| Electrical fields |
|
|
[73][74] | ||||||
| Magnetic fields |
|
|
[75][76] | ||||||
| Ultrasound waves |
|
|
[77][78] | Exparel®(C2H4O) | FDA: 2011 EMA: 2021nC39H76NO10PNa |
Bupivacaine liposome injectable suspension.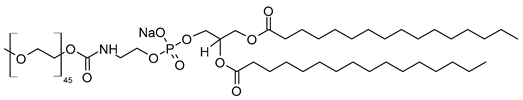 |
|||
| Cholesterol, DPPG, DEPC. | Postsurgical regional analgesia. | Pacira Pharmaceuticals, Inc. (Parsippany, NJ, USA) | |||||||
| Transferrin | Resveratrol | Cationic | |||||||
| Onivyde® | EMA: 2016 | Irinotecan sucrosofate in PEGylated liposomes. | DSPC, cholesterol, MPEG-2000-DSPE. | Metastatic adenocarcinoma of the pancreas. | Laboratoires Servier (Servier) (Suresnes, France) | 1,2-dioleoyl-3-trimethylanmmonium-propane (chloride salt) (CAS No.: 132172-61-3) |
DOTAP | C42H80NO4Cl | 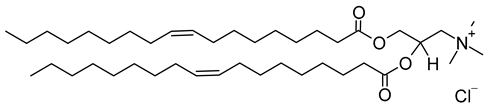 |
| Vyxeos® | FDA: 2017 | Cytarabine and daunorubicin liposome injection. | DSPC, DSPG, cholesterol. | Acute myeloid leukemia. | Jazz Pharmaceuticals plc (Dublin, Ireland) | 1,2-di-O-octadecenyl-3-trimethylammonium propane (chloride salt) (CAS No.: 104872-42-6) |
DOTMA | C42H84ClNO2 | 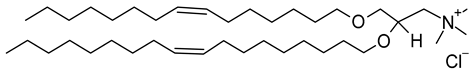 |
| Zwitterion | |||||||||
| 1,2-dimyristoyl-sn-glycero-3-phosphocholine (CAS No.: 18194-24-6) |
DMPC | ||||||||
| Arikayce® | FDA: 2018 EMA: 2020 |
Amikacin liposome inhalation suspension. | Cholesterol, DPPC. | Non-tuberculous mycobacterial (NTM) lung infections. | Almac Pharma Services Ltd. (Athlone, Ireland) | C36 | |||
| Zolsketil® | EMA: 2022 | Doxorubicin in PEGylated liposomes. | MPEG 2000-DSPE, HSPC, cholesterol. | Ovarian neoplasms, sarcoma, Kaposi, multiple myeloma. | Accord Healthcare S.L.U. (Barcelona, Spain) | H72NO8P | 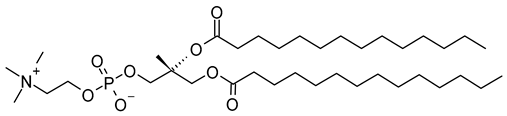 |
||
| 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (CAS No.: 63-89-8) |
DPPC | C40H80NO8P | 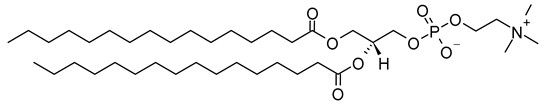 |
||||||
| 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine (CAS No.: 1069-79-0) |
DSPE | C41H82NO8P | 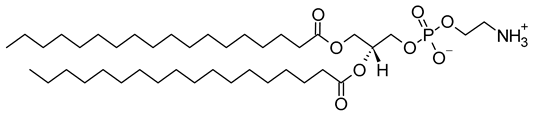 |
||||||
| L-α-phosphatidylcholine, hydrogenated (soy) (CAS No.: 97281-48-6) |
HSPC | C42H84NO8P | 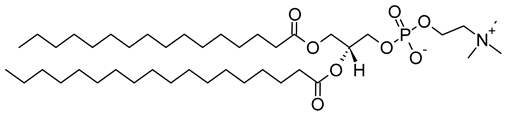 |
||||||
| 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (CAS No.: 26853-31-6) |
POPC | C42 | |||||||
1.2. Targeting Mechanisms of Liposomes
The tumor-targeted delivery of liposomes can be achieved by two main targeting mechanisms: passive and active targeting.
1.2.1. Passive Targeting of Liposomes
In the passive targeting mechanism, liposomes are transported through the tumor interstitium to the target cells through capillary fenestrations and channels through passive diffusion or convection [22]. The tumor angiogenesis induces irregularities in endothelial cells with different pore sizes, ranging from 100 nm to 2 μm [23]. The differences in pore sizes and size distributions between the tumor microvasculature of endothelial cells and the tighter structures of normal cells make liposomes more easily accessible to the cancerous sites. Additionally, liposomes exploit the enhanced permeability and retention (EPR) effect for tumor targeting by improving the amounts of drugs delivered to tumor sites [23]. In order to passively target liposomes to tumor cells, liposomes should possess some physical and structural characteristics, such as the following: (1) the size of the liposomes should be in the range of 10–100 nm; (2) they should carry a neutral or anionic charge in order to avoid renal elimination; and (3) they should be protected from the RES [22][24][22,24].
1.2.2. Active Targeting of Liposomes
Site-specific drug delivery is a method of targeting drugs to specific sites in a manner that increases their therapeutic indexes and reduces their possible side effects and toxicities [22][24][22,24]. Liposomes can reach tumor sites passively through the EPR effect [23], while surface-modified (or surface-engineered) liposomes act by binding to specific receptors overexpressed by cancer cells, such as epidermal growth factor receptor (EGFR), folate receptor (FR), transferrin receptor (TFR), and other receptors (
| Thin-film hydration | |||
| [ | |||
| 32 | |||
| ] | |||
| Mannose | |||
| Chlorogenic acid | Thin-film hydration | [ | 33] |
| cRGD | microRNA | Thin-film hydration | [34] |
| Cationic Liposomes | |||
| Transferrin | Doxorubicin | Ethanol injection | [35] |
| mAbs (Herceptin) | Curcumin | Thin-film hydration | [36] |
| Aptamer (AS1411) | Paclitaxel and siRNA | Thin-film hydration | [37] |
| H | |||
| 82 | |||
| NO | 8 | P | 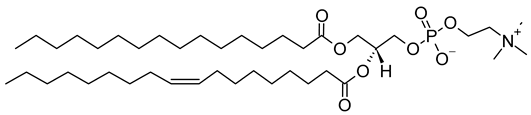 |
| 1,2-Dioleoyl-sn-Glycero-3-phosphocholine (CAS No.: 4235-95-4) |
DOPC | C44H84NO8P | 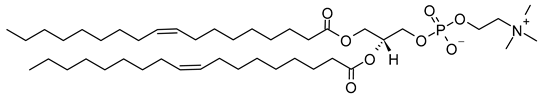 |
| 1,2-Distearoyl-sn-glycero-3-phosphocholine (CAS No.: 816-94-4) |
DSPC | C44H88NO8P | 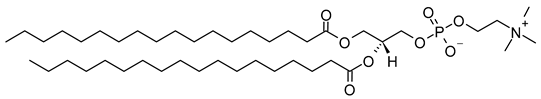 |
| Photoswitchable Lipids | |||
| 1-stearoyl-2-[(E)-4-(4-((4-butylphenyl)diazenyl)phenyl)butanoyl]-sn-glycerol (CAS No.: 1985595-31-0) |
18:0-PhoDAG | C41H64N2O5 | 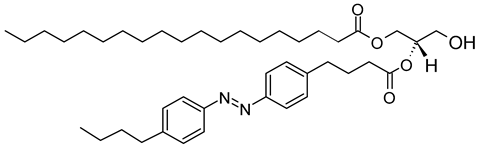 |
| N-[(E)-4-(4-((4-butylphenyl)diazenyl)phenyl)butanoyl]-D-erythro-sphingosylphosphorylcholine (CAS No.: 2260670-56-0) |
Azo SM | C43H71N4O6P | 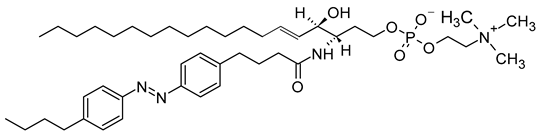 |
| 1-stearoyl-2-[(E)-4-(4-((4-butylphenyl)diazenyl)phenyl)butanoyl]-sn-glycero-3-phosphocholine (CAS No.: 2098674-45-2) |
18:0-azo PC | C46H76N3O8P | 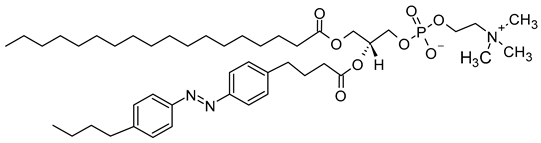 |
Phospholipids play key roles in the stability of liposomes in the systemic circulation, liposomal encapsulation, drug loading efficiency, and drug release at target sites. For instance, the use of 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) in the preparation of liposomes resulted in a higher drug encapsulation efficiency compared to 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC). This was mainly due to the lengthy fatty acid chain of DSPC as well as the rigidity of the acyl chains of DSPC [12]. Furthermore, the use of n-(methoxypolyethylene glycol 5000 carbamoyl)-1,2-dipalmitoyl-sn-glycero-3-phosphatidylethanolamine, monosodium salt (MPEG-5000-DPPE-Na) prolonged the blood circulation time of liposomes, owing to the additional steric hindrance of MPEG-5000-DPPE-Na, which reduced the liposomal uptake by the reticuloendothelial system (RES) [13]. Additionally, 1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol, sodium salt (DPPG-Na) exhibited fusogenic activity that improved the ability of liposomes to cross the cell membrane [14]. In general, liposomes can enter cells either by endocytosis (i.e., the process of capturing liposomes from outside by engulfing them with the cell membrane) or via exocytosis membrane fusion (i.e., the process where two phospholipid bilayers merge into a single continuous bilayer). Anionic liposomes showed faster endocytosis that enhanced their intracellular uptake, while fusogenic liposomes demonstrated an ability to fuse and penetrate the cell membrane [2][9][2,9]. Fusogenic liposomes are a particular type of liposome that are capable of causing fusion with biological membranes, thereby improving cell-type-specific delivery and therapeutic efficacy. They are mainly composed of phospholipids, such as dioleoyl-phosphatidylethanolamine (DOPE) and cholesteryl hemisuccinate (CHEMS) [15]. On the other hand, the use of cholesterol in liposomes aims to provide additional rigidity to the bilayer system in order to enhance liposome stability by increasing the molecular packaging of phospholipid molecules, prompting drug retention inside the bilayer system, and reducing the permeability of phospholipid bilayers [16]. In fact, cholesterol does not form bilayers by itself but will dissolve readily in the phospholipid–water bilayer system [10]. The unique feature of liposomes is their ability to compartmentalize and encapsulate both hydrophilic and hydrophobic drugs. This unique feature, along with biodegradability, biocompatibility, safety, non-toxicity, and targetability, made liposomes very attractive nanocarriers to maximize drug delivery and activity [2][9][2,9].
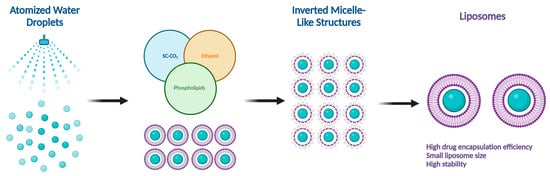
There are several methods for the preparation of liposomes (Figure 1) [2][9][2,9], such as thin-film hydration (or thin-layer evaporation), reverse-phase evaporation, double emulsification, ether injection, ethanol injection, and detergent removal. However, all these techniques are used for lab-scale production and mostly require the use of organic solvents in high concentrations and ratios. Moreover, they exhibit difficulty in controlling size and intercalation efficiency. Supercritical carbon dioxide (SC-CO2) (Figure 2) is a novel technique suitable for the large-scale production of liposomes with the advantages of high encapsulation efficiency and uniform particle size distribution without the necessity of post-formation processes such as sonication or extrusion [17][18][17,18]. In SC-CO2, CO2 is premixed with lipids and then enters a chamber with atomized water droplets. As a result of the high diffusion ability of CO2 and the reduced viscosity of the solution, lipids would coat water droplets at higher rates, resulting in inverted micelle-like structures, which are further stabilized by another layer of lipids placed at the bottom of the chamber [19].
mAbs: Monoclonal antibodies; MM-302: An anti-HER2 mAb; Sortagged anti-EGFR: Sortase-A mediated CH–LPETG–mAb; cRGD: Cyclic arginine-glycine-aspartic acid peptide; RNA: Ribonucleic acid; Herceptin: An anti-HER2 mAb; Aptamer (AS1411): A guanosine-rich oligonucleotide aptamer; siRNA: Small interfering RNA.
1.3. Functionalized Liposomes
Functionalized liposomes include long-circulating PEGylated liposomes, targeting ligand-functionalized liposomes, and stimuli-responsive liposomes (Figure 5).
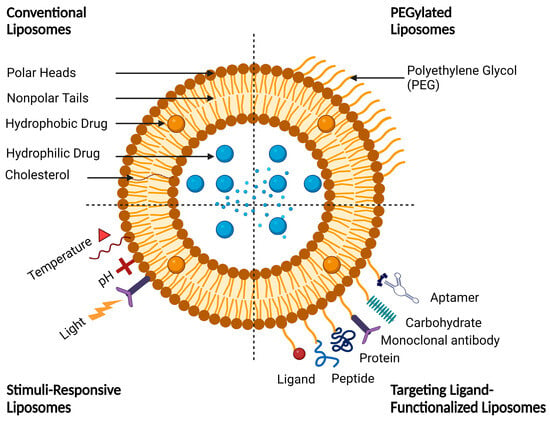
Figure 5.
A schematic representation of conventional and functionalized liposomes.
1.3.1. Long-Circulating PEGylated Liposomes
Effective targeting requires the design of smart drug delivery systems with a long circulatory half-life (i.e., to remain in the bloodstream for longer periods of time), which means that liposomes must evade uptake by RES organs. This unique character can be imparted onto liposomes by coating their surfaces with polymers that suppress opsonization by plasma proteins [38]. These liposomes are commonly known as stealth or long-circulating liposomes, owing to their stealth properties that make them resistant to recognition and degradation by enzymes and immune systems [38][39][38,39]. The most commonly used polymer to prevent liposome opsonization is polyethylene glycol (PEG). The process of PEG attachment to liposomes is called PEGylation [39]. Of note, PEG, commercially known as macrogol, is a hydrophilic, biodegradable polymer with a general formula of H(OCH2CH2)nOH, where n is the number of oxyethylene groups + 1 [40]. There are different grades of PEG available on the global pharmaceutical market, such as PEG-400, PEG-1500, PEG-4000, PEG-6000, and PEG-20000, that differ in their molecular weights. PEGs with molecular weights of 1000 and higher are solid grades and range in their consistencies from pastes to waxy flakes, while PEGs with molecular weights below 1000 are viscous liquid grades [40]. The adsorption of PEGs onto the liposomal surfaces leads to enhanced blood circulation time, reduced RES uptake, increased biodistribution and target accumulation, and enhanced formulation stability [41][42][41,42]
2. Mechanisms of Light-Triggered Drug Release from Liposomes
Light-triggered mechanisms that can be exploited to release encapsulated drugs from liposomes are photoisomerization, photocleavage (photo-oxidation), surface plasmon resonance absorption (photothermal activation), photochemical hydrophobicity change (photochemical activation), and photo-crosslinking and de-crosslinking (Figure 6).
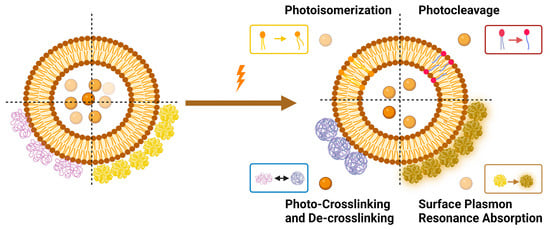
Figure 6.
Light-triggered mechanisms used in triggering drug release from liposomes.
2.1. Photoisomerization
Photoisomerization is a photo-induced isomerization process from one isomeric form to another (i.e., cis (Z)- to trans (E)-isomer). It is worth mentioning that trans (E)-isomers are more stable and lower in energy than cis (Z)-isomers due to no electrical repulsion since the two larger groups are as far away as possible from each other, while in the case of cis (Z)-isomers, the two larger groups bump into one another, resulting in an electrical repulsion [79]. When photo-responsive molecules are irradiated with UV light, they undergo conformational changes from trans- to cis-isomers. These conformational changes make the structural integrity of liposomes more permeable, owing to the steric hindrance as well as the increased polarity of cis-isomers [79]. The transition from trans- to cis-isomers can be triggered by UV light irradiation at wavelengths ranging from 320 to 350 nm, and the reverse transition can be triggered by visible light irradiation (400–450 nm) or by heat.
Azobenzene, spiropyran, and diarylethene are the most commonly used photoswitches in photoisomerization-based drug release [80][81][80,81]. Azobenzene undergoes a UV-light-induced double-bond isomerization to its metastable Z-isomer, which is characterized by being shorter in length, bent, twisted, and more hydrophilic than the E-isomer. Spiropyran (carrying a neutral charge) undergoes a UV-light-induced ring-opening reaction to its zwitterionic metastable form, which is commonly known as merocyanine and is characterized by being more hydrophilic. While diarylethene undergoes a UV light (6π) electrocyclization and ring-closing reaction to its thermally stable isomer, which is characterized by being conjugated and rigid in structure, the ring-closed isomer can be reopened again using visible light (Figure 7) [80][81][80,81].
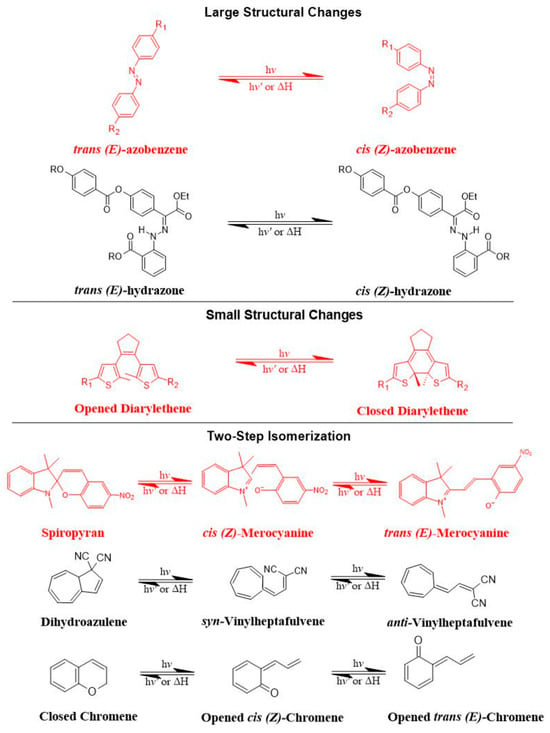
Figure 7.
The most common photoswitches and their photoisomerization reactions.
There are preferred application areas for each photoswitch. For example, azobenzenes are superior photoswitches when large structural and geometrical changes are required [82]. Complementary to azobenzenes, diarylethenes show small structural and geometrical changes but large electronic changes upon photochemical interconversion between the ring-opened and -closed structures [83]. Spiropyrans offer unique properties with respect to ring-opening and -closing isomerization, owing to their molecular dipole moments, which increase during photoconversion processes from ring-closed to -opened structures (Figure 7) [84].
The mechanism of trans-cis photoisomerization has been used to induce drug release from light-responsive liposomes. As an interesting example of the photo-response mechanism of the azobenzene photoswitch in liposomes, Li et al. [85] developed a novel liposomal curcumin formulation with photoswitching properties, owing to the presence of 4-butylazobenzene-4-hexyloxy-trimethyl-ammoniumtrifluoro-acetate (BHA) as a photo-responsive reversible switch. The azo-group of BHA was capable of undergoing a reversible trans-cis isomerization under UV and visible light irradiation. BHA-curcumin-liposomes, abbreviated BHA-cur-lipo, were prepared using the thin-film hydration method along with the SC-CO2 technique. The percent encapsulation efficiency (EE%) of curcumin in BHA-cur-lip was ~88%. Curcumin was released from BHA-cur-lipo under UV light irradiation; ~90% curcumin was released within 6 h. BHA embedded in the liposomal bilayer was able to isomerize under UV light irradiation, and the isomerization process was capable of being repeated multiple times. The isomerization of BHA in the liposomal bilayer could be used as a switch for precisely controlled, on-demand drug release.
As an interesting example of the photo-response mechanism of spiropyran photoswitch in liposomes, Zhang et al. [86] developed photo-responsive liposomes composed of spiropyran-containing triazole-phosphatidylcholine (SPTPC). SPTPC was synthesized through a copper-catalyzed azide alkyne cyclo (CuAAC)-addition reaction. In an aqueous solution, SPTPCs self-assembled into vesicles due to the presence of phosphatidylcholine (PC), and then a spontaneous isomerization of spiropyran-to-merocyanine (SP-to-MC) occurred, resulting in the co-occurrence of liposomes and fibers. The switching from spiropyran (SP) to merocyanine (MC) isomeric form induced a reversible transition between these molecular structures. Additionally, the authors studied the self-assembly properties of SPTPCs and photoinduced liposome–fiber assembly-transition and concluded that (1) the presence of MC allowed for additional intermembrane interaction during self-assembly, and (2) the driving force for the assembly-transition was the MC-stacking effect. Exposure to UV light at 365 nm induces switching from SP to MC isomeric form, where the planar structure and confinement of MC lead to enhanced MC-stacking. The MC-stacking effect had some advantages and drawbacks, such as disrupting the hydrophobic phase in the lipid bilayer and permitting the liposome-to-fiber transition; otherwise, the MC-stacking blocked the switching of MC to SP and caused an incomplete isomerization recovery from MC to SP during fiber-to-liposome recovery. Therefore, a fatigue of SP was observed during the liposome-to-fiber transition cycle. To suppress the MC-stacking effect and minimize the intermolecular interaction, a photo-inert triazole-phosphatidylcholine (TPC) was subsequently added to make TPC/SPTPC-liposomes, which showed better recovery kinetics. The active photoadaptation behavior of TPC/SPTPC-liposomes confirmed the disturbance of the lipid bilayer through the formation of MCTPC-enriched phases in the lipid bilayer. To conclude, the reversible liposome-to-fiber assembly transition of SPTPC is a promising and potential candidate for adaptive assembly systems.
As an interesting example of the photo-response mechanism of diarylethene photoswitch in liposomes, Liu et al. [87] synthesized a novel amphiphilic photoswitchable fluorescent probe of liposomes, namely, PEGylated perylenemonoimide-dithienylethene, abbreviated PEG-PMI-DTE, that exhibited excellent photochromic reversibility, fluorescence switching, and fatigue resistance under UV and visible light irradiation. The fine nanostructures of liposomes (MLVs, LUVs, and SUVs) were able to be observed directly under a super-resolution optical microscope with the use of amphiphilic photoswitchable fluorophore as a staining agent, with an optical resolution of 30 nm. This research offers a new type of optical probe and an optical approach to investigating nanostructures using photoswitchable fluorescent probes in super-resolution imaging.
2.2. Photocleavage (Photo-Oxidation)
Photocleavage is a photo-induced bond cleavage through photosensitized oxidation that can be achieved when the photosensitizer and oxygen are proximate to the oxidizable lipid in liposomes, which are characterized by having a lipid segment sensitive to singlet oxygen (1O2) produced by the photosensitizer [88]. The photocleavage release mechanism from liposomes occurs mainly through lipid photo-oxidation [89], which leads to membrane destabilization, disruption, and subsequently drug release [89]. Briefly, when liposomes are irritated with light, the photosensitizer will absorb photos, leading to an excited triplet state. This generates reactive oxygen species (ROS) that can be either in the form of radicals (hydroxyl (HO•) and superoxide (O2•)) or non-radicals (1O2). Singlet oxygen (1O2) is a highly reactive oxidant with low stability and a short half-life. It can oxidize different cellular constituents, such as nucleic acids, lipids, and proteins [88][89][90][88,89,90]. Its tendency to induce toxicity can be precisely controlled.
The mechanism of photocleavage was explored through photodynamic therapy (PDT). PDT is a light-based cancer therapy that uses light to activate photosensitizers, leading to the generation of ROS, or 1O2, which are highly reactive oxidants that can mediate damage to tumor cells or tissues. The effectiveness of PDT depends on several factors [91][92][91,92], such as (1) the type of photosensitizer, (2) the intensity of light, (3) the route of administration, (4) tumor type, size, and location, and (5) the concentration of dissolved cytoplasmic oxygen. The ideal photosensitizer should be [91][93][94][91,93,94] (1) safe, effective, and non-toxic; (2) a water-soluble compound; (3) pharmacologically inactive in the absence of a light source; (4) highly specific and selective; (5) have an absorption spectrum preferably between 650 nm and 800 nm; and (6) be rapidly metabolized to metabolites and discharged from the human body. Photosensitizers are categorized into two main classes: porphyrin photosensitizers and non-porphyrin photosensitizers [94]. Three generations of porphyrin photosensitizers exist. First-generation porphyrin photosensitizers include hemaporphyrins, which have several drawbacks that limit their therapeutic use, such as (1) chemical instability issues; (2) poor tissue penetration; (3) activation with light below 650 nm; (4) skin hypersensitivity reactions; (5) long half-life; and (6) low elimination rates [94]. Second-generation porphyrin photosensitizers include metalloporphyrins, porphycenes, purpurins, chlorins, and protoporphyrins [94]. Second-generation porphyrin photosensitizers have been approved by the FDA and EMA for the treatment of cancer. For example, 5-aminolevulinic acid (ALA) and methyl aminolevulinate (MAL, Metvix®, Galderma, Lausanne, Switzerland) are precursors of protoporphyrin IX, which absorbs at 630 nm. They are approved by the FDA for the treatment of prostate, bladder, and colon cancers [20]. Meta-tetrahydroxy-phenyl chlorine (m-THPC, Temoporfin, Foscan®, Biolitec Pharma, Vienna, Austria) absorbs at ~652 nm and is approved by the EMA for the treatment of biliary and pancreatic cancers [21]. Verteporfin (Visudyne®, Novartis, Basel, Switzerland), a benzo-porphyrin derivative, absorbs at 690 nm and is approved by the FDA for the treatment of gastric cancer [20]. Coupling the second-generation porphyrin photosensitizers with biologically targeting molecules, such as carbohydrates, peptides, or antibodies, resulted in the third-generation porphyrin photosensitizers, which displayed high selectivity and specificity with minimal adverse effects [94]. Non-porphyrin photosensitizers include psoralens, anthracyclines, chalcogenopyrylium dyes, cyanines, and phenothiazinium dyes [94]. Although all of the above advantages of PDT photosensitizers in the treatment of cancer exist, they are still suffering from serious drawbacks and limitations, such as poor biodistribution and cellular uptake of hydrophobic photosensitizers, difficulty in applying PDT to deeper tumor tissues, and low sensitivity and selectivity towards some cancer cells [91][92][93][94][91,92,93,94]. Therefore, PDT is only effective and suitable for treating superficial skin tumors.
Since the use of photosensitizers often causes serious skin hypersensitivity reactions, the encapsulation of photosensitizers into nanocarrier systems, such as liposomes, will overcome these problems. Of note, Sun et al. [95] developed anticancer liposomal chemophototherapy (CPT) using a bilayer-loaded photosensitizer and the anti-cancer drug cabazitaxel (CTX). Cabazitaxel-loaded porphyrin–phospholipid liposomes, abbreviated CTX-PoP-Lip, were prepared using the hot ethanol injection method in order to encapsulate the hydrophobic CTX within the lipid bilayers. Cholesterol and PEG-lipid were added to enhance liposomal stability and permeation. The EE% of CTX in CTX-PoP-Lip was ~60%, and the percentage of loading capacity (LC%) was ~2%. Morphologically, CTX-PoP-Lip showed spherical, unilamellar vesicles with a diameter size of ~100 nm. CTX-PoP-Lip showed an optical absorption peak similar to PoP-Lip without CTX, with a characteristic PoP peak apparent at 420 nm (for the PoP Soret band) and 675 nm (for the PoP Q-band). Upon excitation at 675 nm, a fluorescence peak was observed for both CTX-PoP-Lip and PoP-Lip. Without PoP, CTX-Lip had no fluorescence. Over 3 months of storage under 4 °C, CTX-PoP-Lip displayed good colloidal stability in terms of particle size, polydispersity, and zeta potential. Moreover, CTX showed good photochemical stability under laser irradiation. Remarkably, the combination of CTX-PoP-Lip with laser treatment showed a positive tumor inhibition therapeutic effect in comparison with PDT alone or chemotherapy alone.
Lipid–porphyrin conjugates are novel, promising carriers for drug delivery with multifunctional and light-triggered release properties. These compounds are able to self-assemble into liposome-like structures to form porphysomes that consist of porphyrin–lipid conjugates generated by acylation reactions between phospholipids and chlorophyll-derived porphyrin analogues [96]. As a result of this supramolecular self-assembly, the porphysome bilayer contains an extremely high porphyrin packing density, resulting in extreme self-quenching of Pyro fluorescence [96]. Upon NIR irradiation, the energy absorbed is released primarily as heat, generating temperatures in magnitudes comparable to those of inorganic nanoparticles. This large heat generation makes porphysomes a very promising photothermal sensitizer [97]. Moreover, the structure-dependent self-quenching properties of porphysomes enable fluorescence imaging upon the disruption of the porphysome bilayer. As porphysomes are captured by cells via endocytosis, the structure of porphysomes is disrupted, and fluorescence is greatly increased. This activatable fluorescence indicates that the porphysomes have reached the desired target site [96][97][96,97]. On the other hand, the porphyrin rings offer chelating sites for metal atoms, thus making porphysomes potential candidates for the development of multimodal imaging contrast agents. Alongside these photosensing properties, porphysomes possess liposome-like characteristics with improved photophysical properties [98]. For instance, porphysomes can be actively targeted at tumor cells by modifying their surfaces through any of the active targeting strategies used by conventional liposomes, as previously described [99].
As a promising example of these self-assembled structures, Cressey et al. [100] developed novel liposome-like assemblies composed of newly synthesized phospholipid–porphyrin conjugates bearing either pheophorbide-a (Pheo-a) or pyropheophorbide-a (Pyro-a) photosensitizers. These conjugates presented different alkyl chain lengths in the sn-2 position and were linked to either pheophorbide-a (PhxLPC) or pyropheophorbide-a (PyrxLPC) via amide coupling (Figure 8). These liposome-like structures were composed of lipid–porphyrin conjugates made of cholesterol, a phospholipid–porphyrin conjugate, and DSPE-PEG-2000 in a ratio of 47.5:47.5:5 mol% and were prepared using the thin-film hydration method, followed by the extrusion of the vesicles. Interestingly, all phospholipid–porphyrin conjugate assemblies showed similar absorbance and fluorescence, with higher fluorescence quenching for Pyro-a conjugates. Formulations containing the phospholipid–porphyrin conjugate with the longest linker displayed higher stability than those with a shorter one. This could be due to the deeper embedment of the porphyrin core inside the lipid matrix. Based on these results, the authors selected Pyr3LPC and Ph3LPC liposomes as promising candidates for in vitro studies. Another very promising example is Massiot et al. [101], who designed photo-triggerable liposomes based on the lipid–porphyrin conjugate and cholesterol combination. First, they synthesized a new lipid–porphyrin conjugate, termed PhLSM, by coupling pheophorbide-a (Pheo-a) with egg lyso-sphingomyelin (Figure 8). The pure PhLSMs were able to self-assemble into liposome-like structures, but they were highly unstable due to the mismatch between the length of the alkyl chain in the sn-1 position and the adjacent porphyrin. Stable PhLSM lipid bilayers were obtained by mixing PhLSMs with cholesterol, which is known to exhibit a complementary geometrical packing parameter. Based on these observations, the authors prepared stable liposomes encapsulating a hydrophilic fluorescence probe in the aqueous core. The prepared liposomes showed light-triggered cargo release in an ON/OFF fashion, which was attributed to their photothermal conversion. In addition to the light-triggered cargo release property and phototoxic photothermal effect, the prepared liposomes showed a markedly high photothermal conversion efficiency and photostability.
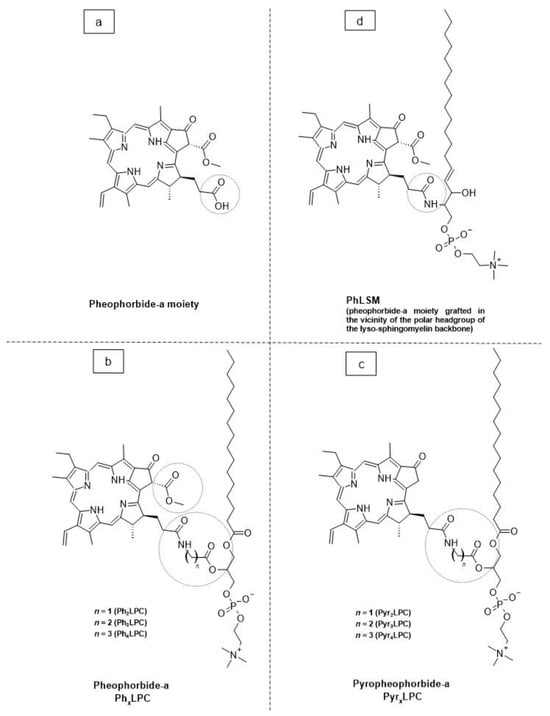
Figure 8. The chemical structures of (a) pheophorbide-a; (b) pheophobide-a PhxLPC; (c) pyropheophorbide-a (PyrXLPC); and (d) PhLSM.
2.3. Surface Plasmon Resonance Absorption (Photothermal Activation)
Photothermal approaches involve the conversion of light into heat to induce liposomal membrane permeabilization. Some metals exhibit unique optical properties, such as dielectric function, reflectivity, and electron energy loss function, when present in the form of nanostructures as nanoparticles or entrapped inside the nanocarrier systems as nanoparticle-loaded liposomes [102]. Metallic nanostructures are highly attractive multifunctional nanoplatforms, owing to their unique size- and shape-dependent properties [102]. One of the most interesting characteristics of metallic nanostructures is their optical properties, which are strongly dependent on particle size and shape [102]. For example, bulk gold metals look yellowish in reflected light, but thin gold films look blue in transmission. This characteristic blue color gradually changes to orange through several tones of purple and red as a result of reducing particle size to ~3 nm. These changes are likely to account for the surface plasmon resonance (SPR) [102], which is defined as the frequency or wavelength at which conduction electrons oscillate with regard to the alternating external electric field (Figure 9). The optical properties of metallic nanostructures are controlled by the collective oscillation of conduction electrons, resulting from the interaction with the electric field of the incident light, owing to the presence of free conduction electrons [103]. The electric field of the incoming radiation creates a strong dipole electric field inside the metallic nanostructures. A restoring force in the metallic nanostructures attempts to compensate for this difference, resulting in a unique resonant wavelength [103].
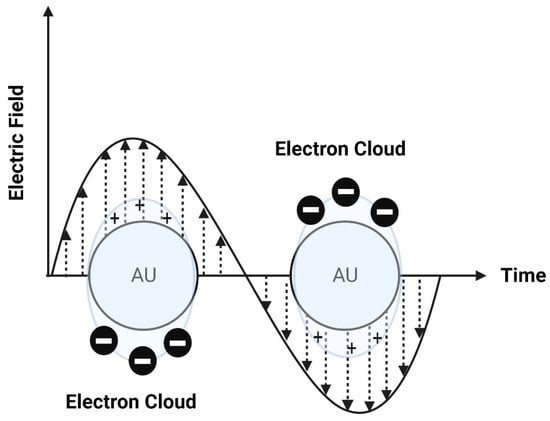
Figure 9.
A schematic diagram of surface plasmon resonance (SPR).
The SPR frequency and intensity of metallic nanostructures are dependent on the electron charge density, which is primarily affected by several factors, such as size, shape, structure, composition, and the dielectric constant of the surrounding environment.
Interestingly, Rubio-Camacho et al. [104] synthesized stable gold nanoparticles on the surface of DPPC thermosensitive liposomes, termed AuNPs@DPPC, resulting in the formation of nanohybrids with an on-demand plasmon mode in the visible/NIR region and with good photothermal conversion efficiency. The AuNPs@DPPC nanohybrids retained the physical properties of DPPC thermosensitive liposomes without altering either the liposome fluidity or the hydration degree of the lipid bilayer. The AuNPs@DPPC nanohybrids showed good light-to-heat conversion properties upon irradiation in the NIR region. These nanohybrids represented highly attractive and promising candidates in light-mediated therapies, such as NIR-light-controlled drug delivery. As an interesting example of gold nanoparticle–drug conjugates in liposomes, Li et al. [105] synthesized vincristine sulfate-conjugated gold nanoparticles incorporated into liposomes as a promising light-responsive hybrid nanocarrier system with enhanced antitumor efficiency. Gold nanoparticles were synthesized by reducing tetrachloroaurate, using trisodium citrate as the reducing agent. The amount of trisodium citrate used in the synthesis of gold nanoparticles was 1% to prepare uniform size-controlled nanoparticles. The resulting gold nanoparticles had a particle size of 17 nm. The conjugation of gold nanoparticles with vincristine sulfate was achieved via ionic bonding, since vincristine sulfate is positively charged while the citrate-capped gold nanoparticles are negatively charged. The highest EE% was achieved when the vincristine–gold nanoparticle molar ratio was 6:100. The conjugates were incorporated into liposomes using film dispersion to yield nanoparticles of 113.4 nm with UV light-responsive, controlled release properties. Interestingly, UV irradiation also considerably increased intracellular drug release, cytotoxicity, and apoptosis in HeLa cells. In vivo studies in tumor-bearing nude mice showed that the therapeutic efficacy of vincristine was enhanced after exposure to UV light, with a relatively high tumor inhibition rate and low toxicity. The accumulation of the drug selectively at the tumor site (by the EPR effect of liposomes), together with light-responsive controlled release, represented an important step forward in tumor-targeting drug delivery.
2.4. Photochemical Hydrophobicity Change (Photochemical Activation)
Amphiphilic block copolymers have a relatively high potential to produce nanostructures (either micelles or vesicles) via self-assembly in suitable solvent systems [103]. Polymeric micelles are thermodynamically stable when the concentration of polymers is above the critical micelle concertation (CMC) value. If the concentration of polymers is below the CMC value, micelles will disintegrate, dissolve, and release their payloads. Thus, in such cases, polymeric micelles are thermodynamically instable [103]. Therefore, various techniques were developed and applied to improve the thermodynamic stability of polymeric micelles. Foremost among these techniques is a photochemical activation method based on changing the hydrophobicity of molecules [103][106][103,106]. Briefly, this mechanism depends on increasing the CMC value and dissolving the micelles by converting the amphiphilic polymers to more hydrophilic forms, thus providing a controlled drug release [103][106][103,106]. Interestingly, light-responsive chromophores, such as azobenzene, spiropyran, diarylethene, and their derivatives, can be incorporated inside the micellar cavity, where NIR can be used to induce chemical transformation to a more hydrophilic form [106]. Most light-responsive chromophores can absorb UV light; however, NIR is more suitable for biomedical applications owing to its capability to penetrate deeply into tissues (up to 10 cm) with a low potential for tissue damage [106]. Self-assembled polymeric micelles are used as amphiphilic particulate emulsifiers for controllable Pickering emulsions. Pickering emulsions have been developed unprecedently in drug delivery. However, engineering tunable Pickering emulsions with the capability of responding to light still remains very challenging. Interestingly, Zhao et al. [107] designed a photo-controllable nanocarrier system to control the amphiphilicity of Pickering emulsifiers using a β-cyclodextrin-grafted alginate polymer and an azobenzene derivative. Briefly, a biocompatible alginate polymer grafted with β-CD (via the Ugi reaction), abbreviated Ugi-Alg-CD, was first synthesized and used as an amphiphilic macromolecule surfactant host. Then, azobenzene coupled with polyethylene glycol (Azo-PEG) was prepared and used as a guest molecule. By coupling Ugi-Alg-CD with Azo-PEG, a stable Pickering emulsion was successfully fabricated. The photoisomerization of a host–guest complex between β-cyclodextrin and an azobenzene derivative was customized to regulate the polarity of the microenvironment. Interestingly, the photoactivatable emulsifier based on supramolecular self-assemblies was able to undergo destabilization of O/W emulsions by changing the amphiphilic balance of host–guest assemblies at the O/W interface under UV light irradiation, resulting in phase separation. The analysis of the microstructures of self-assemblies at the O/W interface during the demulsification process indicated that the reversible light-triggered trans-cis isomerization of Azo-PEG likely resulted in the regulation of the hydrophilic-hydrophobic balance of supra-amphiphilic polymer emulsifiers. This photochemical strategy opened the door to developing novel photo-responsive nanocarrier systems for various biomedical applications.
Polymersomes are biomimetic cell membrane-like bilayer vesicles that are self-assembled stepwise from amphiphilic block copolymers. They are analogous to liposomes but with outstanding properties, such as a higher chemical stability towards oxidation and hydrolysis reactions and a greater resistance to mechanical deformation processes within the human body like bending and stretching (i.e., resistance to high shear rates of blood circulation and deformations during blood flow through microvessels) or cellular processes (e.g., division and fusion) [108]. Moreover, other properties, such as composition, size, shape, and surface chemistry, resulted in increased EE% and LC% (i.e., polymersomes have a lower membrane fluidity and higher viscosity due to the presence of amphiphilic block copolymers), which contribute to the low permeability of encapsulated drugs from the inner core of polymersomes to the outer site [108][109][108,109]. Polymersomes can disassemble in response to light to control the release of encapsulated drugs that may also respond to light. Thus, polymersomes can provide spatiotemporal control of drug release. Interestingly, Yamamoto et al. [110] studied the structure–function relationships and photo-release characteristics of different types of photo-responsive polymersomes composed of amphiphilic di-block copolymers. The building blocks of these photo-responsive polymersomes were hydrophobic polymers and poly(ethylene glycol) with photocleavable 2-nitrobenzyl compounds bearing alkyne and maleimide moieties. Interestingly, all polymersomes preserved their hollow structures even after light irradiation. Additionally, polymersomes with a 2-nitrosobenzyl photolysis residue within the hydrophobic shells showed photo-induced drug release after complete photolysis. The authors concluded that the drug release was controlled by photo-induced permeability changes of the hydrophobic shells rather than the decomposition of their molecular structures.
2.5. Photo-Crosslinking and De-Crosslinking
The mechanism of photo-crosslinking-induced drug release occurs through the polymerization of unsaturated bonds located in the hydrophobic domain of the lipid bilayer. When photo-responsive polymerizable moieties are irradiated with light at a specific wavelength, the crosslinking reaction between them causes the shrinkage of the lipid bilayer in the surrounding domain where the photosensitizers are present. This causes bilayer disruption by altering lipid packing; as a result, conformational changes occur, leading to increased membrane permeability and drug release rates [103][106][103,106]. The mechanism of photo-crosslinking was first reported in liposomes by Regen et al. [111]. Liposomes were prepared with a photo-triggerable lipid containing two methacrylated phosphatidylcholine derivatives. The resulting liposomes were more stable than the non-crosslinked type and displayed prolonged blood circulation and enhanced tumor accumulation and retention. More interestingly, Nakamura et al. [112] described the transportation of DNA into liposomes using ultrafast photo-crosslinking. The cohesion of the DNA adsorbed onto the liposomal surface induced transformations in the liposomal structure and allowed photo-triggered, sequence-specific DNA transportation into liposomes. This technique was a useful tool for the specific delivery of nucleic acid drugs.
Reversible photo-decrosslinking is a promising emerging alternative to optimize target-specific drug binding. Photo-decrosslinking was first reported in 2009 by He et al. [113], who formulated a nanogel made with a di-block copolymer (PEO-b-P(MEOMA-co-CMA)) composed of polyethylene oxide (PEO) and a coumarin-containing poly(2-(2-methoxyethoxy)ethyl methacrylate), P(MEOMA-co-CMA). Crosslinking was achieved by using UV light at 310 nm, while de-crosslinking was achieved by irradiating at 260 nm. Recently, Lu et al. [114] developed a photo-responsive microgel that can be reversibly photo-crosslinked and de-crosslinked using UV light of two different wavelengths. This microgel was prepared through the precipitation copolymerization of 2-(2-methoxyethoxy)ethyl methacrylate (MEO2MA), methacrylic acid (MAA), and 7-(2-methacryloyloxyethoxy)-4-methylcoumarin (CMA). The effective crosslinker CMA can be photo-crosslinked through irradiation with UV light at 365 nm and photo-decrosslinked through irradiation with UV light at 254 nm. To understand the photoswitching mechanism, the volume-phase transition temperature (VPTT) was monitored during transitions. The authors concluded that there was a significant change in VPTT that led to a uniform distribution of CMA within the microgel interior. The photo-induced swelling behavior of the microgel was employed to control the release of the anticancer drug doxorubicin. This research study opened the door to developing new hybrid systems of liposome-in-gel as promising carriers for cancer therapy.