Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Anikó Borbás and Version 2 by Sirius Huang.
Viral proteases play a key role in viral replication for all positive single-stranded RNA viruses and some DNA viruses, such as herpesviruses. To treat infections caused by these viruses, proteases are considered excellent drug targets. Protease inhibitors are now routinely used in antiviral therapy for human immunodeficiency virus (HIV) and hepatitis C virus (HCV) infections. Since SARS-CoV-2 Mpro plays a key role in viral replication by cleaving viral polyproteins, inhibition of its catalytic activity represents an attractive therapeutic approach for the treatment of COVID-19.
- viral proteases
- SARS-CoV-2
- non-structural protein (NSP)
- main protease (Mpro)
- 3CL protease
- nirmatrelvir/ritonavir
- electrophilic warhead
- covalent inhibitor
1. Introduction—Viral Proteases as Drug Targets
The genetics and reproduction of viruses differ significantly from what we are used to in eukaryotes in many respects. One important difference is that many viruses, including retroviruses, herpesviruses, flaviviruses and coronaviruses, do not encode functional proteins that are synthesized individually, but rather one or two large polyproteins that are then cleaved by viral proteases into functional proteins [1]. Proteases are a subgroup of hydrolases, enzymes that catalyze hydrolytic reactions. There are many mechanisms of proteolysis; a common method is the use of a nucleophilic group, generated from the side chain of serine (serine proteases) or cysteine (cysteine proteases), which can perform a nucleophilic attack on the partially positively polarized carbonyl carbon atom of the peptide bond. In aspartic proteases, a water molecule bound to aspartic acid in the active site of the enzyme acts as a nucleophile. In the case of serine and cysteine proteases, the nucleophile is generated by the amino acids in the active site of the enzyme. A common system for this is the so-called catalytic dyad or catalytic triad, which consists of two or three amino acids [1][2][1,2]. One of the three amino acids that make up the catalytic triad carries an acidic side chain, e.g., aspartic acid (Asp), the other carries an alkaline side chain, e.g., histidine (His), and the nucleophilic group is formed from the third amino acid, which is serine (Ser) or cysteine (Cys).
In some cysteine hydrolases, histidine and cysteine form a catalytic dyad in the active site; in such enzymes, the role of the third amino acid, Asp, is played by an activated water molecule (Scheme 1). In the first step of the enzyme’s catalytic mechanism, the imidazole ring of histidine as a base deprotonates the thiol group of cysteine, forming a thiolate–imidazolium ion pair. Next, the thiolate group performs a nucleophilic attack on the carbonyl C atom of the peptide bond of its substrate. As a result, the peptide bond is cleaved, the amine terminus of the peptide fragment (R-NH2) is released, while the acyl part forms a thioester with the cysteine, and the histidine is reestablished to its deprotonated form. Finally, the thioester bond of the acylated enzyme is hydrolyzed by an activated water molecule to generate a carboxylic acid group on the remaining substrate fragment (R’-COOH), regenerating the free enzyme.
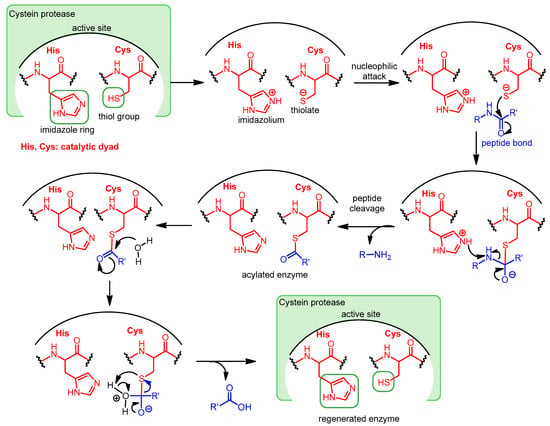
Scheme 1.
Catalytic mechanism of Cys proteases operating with a catalytic dyad.
Viral proteases play a key role in viral replication for all positive single-stranded RNA viruses and some DNA viruses, such as herpesviruses. To treat infections caused by these viruses, proteases are considered excellent drug targets [3]. Protease inhibitors are now routinely used in antiviral therapy for human immunodeficiency virus (HIV) and hepatitis C virus (HCV) infections.
2. Protease Inhibitors as Antivirals
2.1. Protease Inhibitor Drugs for the Treatment of HCV and HIV Infections
The two main groups of protease inhibitors (PIs) currently used in medicine are HIV and HCV protease inhibitors (Figure 1). Hepatitis C virus (HCV) is a small RNA virus that causes hepatic diseases. HCV protease inhibitors, such as asunaprevir, telaprevir, and boceprevir, target the NS3/4A serine protease of the virus (NS stands for “nonstructural” in the name of viral proteins, indicating that the given protein is not a structural protein) [4][5][13,14]. The structures of these protease inhibitors appear to be very different, but all contain at least one peptide (amide) bond (highlighted in red in Figure 1). This is necessary because these inhibitors act by binding to the active site of the enzyme, so they must have a chemical structure similar to the natural peptide substrate, so the inhibitors are peptidomimetics. Some of the inhibitors contain a “warhead” group, which reacts with the enzyme and binds covalently to the active site, thereby inactivating the protease. It is important to note that although inhibitor–protease binding is usually covalent, the inhibition is reversible. The warheads used in the case of serine protease include, for example, an α-ketoamide, boronic acid or α-keto acid group. To learn about the general structure of protease inhibitors, let us take a closer look at boceprevir. In the commonly used nomenclature of protease inhibitors, the positions from the cleavage site towards the C-terminal of the molecule are numbered P1′, P2′, P3′, etc., while the groups towards the N-terminal are P1, P2, P3, etc. [6][15]. Accordingly, there is an α-ketoamide warhead in the P1 position of boceprevir to ensure the covalent inhibitory effect. Other parts of the molecule serve to bind to the enzyme with secondary bonds. The P3 position is “capped” with a carbamide type group.
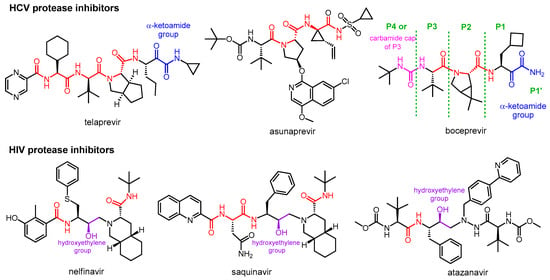
Figure 1.
Structures of viral protease inhibitor drugs.
HIV protease inhibitors are used against the human immunodeficiency virus (HIV), which causes Acquired Immune Deficiency Syndrome (AIDS). HIV protease is an aspartic acid protease that cleaves the peptide bond between a phenylalanine and a proline. Its inhibitors, such as nelfinavir, saquinavir, atazanavir, etc., are peptidomimetics that contain a non-cleavable hydroxyethylene group [7][8][16,17]. HIV PIs are used in highly efficient antiretroviral therapy, which is the standard protocol for treating HIV infection, converting it from a fatal disease to a chronic infection.
2.2. Development and Mechanism of Action of Nirmatrelvir
The structure of nirmatrelvir (PF-07321332) [9][4] from Pfizer can be traced back to a previous Pfizer compound, PF-00835231 [10][18], which was generated to inhibit the main protease of SARS-CoV-1. The zoonotic coronavirus SARS-CoV-1 emerged in China in 2002 and caused an epidemic leading to ~8000 cases and almost 800 confirmed deaths [11][19]. Due to the high similarity (~96% sequence homology) between the main proteases of the two viruses, PF-00835231 can also inhibit SARS-CoV-2 Mpro [9][10][4,18].
The problem with PF-00835231 is that it has very low oral absorption. A development process was started by Pfizer to solve the problem (Figure 2) [9][12][13][4,6,20]. One way to increase the oral bioavailability of a molecule is to reduce the number of hydrogen bond donor groups (HBD) [14][21]. Therefore, the α-hydroxymethyl ketone warhead was changed to benzothiazol-2-yl ketone (highlighted in yellow). Furthermore, the P2 unit was replaced by a pyrrolidine ring (highlighted in green in 1) to get rid of the N–H group, which is an HBD group, to give compound 1. However, these changes eliminated the possibility of H-bonding with amino acid Gln-189 of Mpro, thereby weakening the binding to the target enzyme, which reduced the inhibitory activity. The indole component P3 was replaced with a smaller acyclic sulfonamide unit (highlighted in blue) to fit into the S3 pocket of Mpro, thereby increasing the binding affinity to the enzyme, which increased the inhibitory effect. The antiviral activity of compound 2 obtained through the changes was indeed better than that of compound 1, and its oral bioavailability was higher than that of compound PF-00835231. Subsequently, the P3 cap was replaced with a trifluoroacetamide group (highlighted in green in 3), which further improved antiviral activity, as well as increased metabolic stability and oral pharmacokinetics. Finally, the introduction of the nitrile warhead at the P1′ position (highlighted in green in nirmatrelvir) further enhanced the antiviral activity and oral bioavailability. An additional advantage of the nitrile warhead was that it facilitated the synthesis, since the nitrile-containing compound was more soluble and less prone to epimerization [9][4].
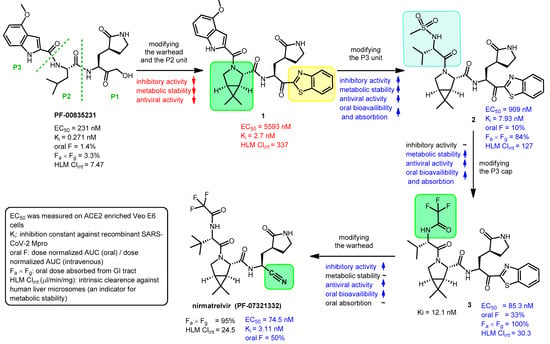
Figure 2. Development of nirmatrelvir. EC50: half-maximal effective concentration, oral F: oral bioavailability (oral fraction) in rat, AUC: area under curve, GI tract: gastrointestinal tract.
During the development process, the activity of the compounds was compared with enzyme inhibitory (Ki) and anti-SARS-CoV-2 (EC50) measurements, and for their pharmacokinetic characterization, oral bioavailability (oral F) and metabolic stability tests were performed [9][4]. Oral bioavailability was assessed in rats and metabolic stability was assessed by measuring intrinsic clearance against human liver microsomes (HLM Clintr). The results of these tests are summarized in Figure 2.
Nirmatrelvir (PF-07321332) is an orally administered covalent inhibitor of SARS-CoV-2 Mpro. As shown in Figure 3A, it has a high structural similarity to boceprevir, but their warheads are different.
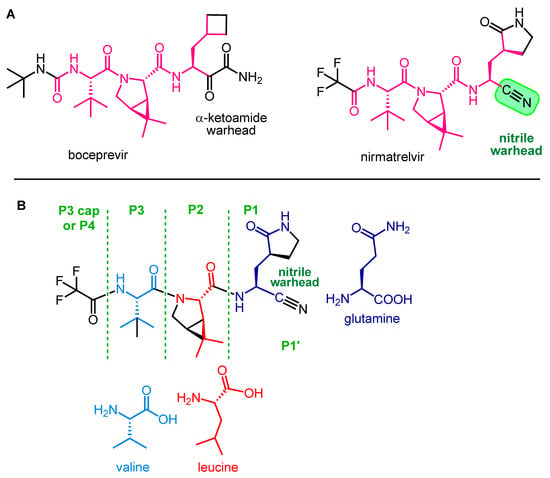
Figure 3.
Structural similarity between boceprevir and nirmatrelvir (
A
) and main structural elements of nirmatrelvir (
B
).
The main structural elements of nirmatrelvir are depicted in Figure 3B. The warhead of the molecule is the nitrile group in the P1′ position, which covalently reacts with the enzyme, while the rest of the molecule, mimicking the natural recognition sequence of Mpro, the tripeptide Val-Leu-Gln (valine-leucine-glutamine), ensures that nirmatrelvir fits into the active site of the enzyme and binds there with secondary bonds [15][22]. The P1 unit is a Gln analog with a cyclic (γ-lactam) side chain that can form H-bonds. It is an advantageous modification because the amide group of native Gln can react intramolecularly with certain types of warheads, rendering the molecule ineffective. This cyclic moiety is more rigid than the native Gln side chain, which may be beneficial in terms of binding to the target enzyme [16][23]. Furthermore, the modification of Gln into a cyclic derivative also helps the synthesis. The P2 group is a dimethylcyclopropyl proline (DMCP) and is a leucine analog that binds primarily to the enzyme at the S2 position through lipophilic interactions. The P3 residue is a tertiary leucine that mimics valine. The N-terminal is capped with a trifluoroacetyl group [9][17][4,24].
The nitrile warhead plays a key role in the protease inhibitory mechanism of nirmatrelvir. It is worth noting that the nitrile group is also used in other drugs (e.g., vildagliptin) and drug candidates (e.g., Cbz-A VLQ-CN) to target serine and cysteine peptidases. Although nitrile is less reactive compared to other warheads (e.g., aldehydes), which can be a disadvantage; however, on the other hand, it provides better selectivity and metabolic stability [16][23].
The covalent interaction between nirmatrelvir and SARS-CoV-2 main protease is shown in Scheme 2. The P1′ nitrile group forms a thioimidate bond with the Cys-145 thiol functional group of Mpro through a Pinner-like reaction. Since the thiol group of Cys-145 is essential for catalyzing the hydrolysis of peptide bonds, the function of the enzyme is blocked [13][18][20,25]. It is important to note that nirmatrelvir alone is ineffective in vivo because it is very rapidly metabolized by the cytochrome CYP3A4 enzyme. For therapeutic use, nirmatrelvir is combined with a CYP3A4 enzyme inhibitor, which acts as a pharmacokinetic enhancer, increasing the bioavailability of nirmatrelvir.
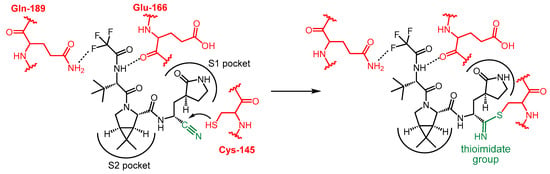
Scheme 2.
Mechanism of action of nirmatrelvir.
2.3. Synthesis of Nirmatrelvir
A synthesis route of nirmatrelvir developed by Pfizer involved the preparation and coupling of the P1 building block 6 and the P2-P3 dipeptide building block 12, with the formation of the nitrile warhead at the P1′ position as the final step (Scheme 3). The production of the P1 building block started from the protected amino acid derivative 4. The amino group of 4 is protected in the form of a carbamate with a tert-butoxycarbonyl (Boc) group, which can be cleaved by acidic hydrolysis, and its carboxyl group is protected in the form of a methyl ester, which can be removed under alkaline conditions. In the first step, compound 4 was treated with methanolic ammonia to cleave the methyl ester to give amide 5, which was Boc-deprotected with hydrochloric acid to form the hydrochloride salt 6. In parallel, N-Boc-t-butylalanine (or N-Boc-3-methylvaline) (7) as the carboxylic acid reactant and compound 8 as the amine reaction partner were coupled to form dipeptide 9, using diisopropylethylamine (DIEA), as the base and O-(7-azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HATU) as coupling agent; the conditions used are common in peptide chemistry. The methyl ester group of 9 was cleaved with LiOH to give compound 10, the Boc group was then removed from the N-terminus with HCl to give compound 11. Ethyl trifluoracetate was used to convert the NH2 group of 11 to acetamide. The obtained compound 12 containing a free carboxyl group was ready for coupling with compound 6. Peptide coupling was performed using DIEA as base, 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDCI) as coupling agent, in the presence of 2-hydroxypyridine 1-oxide (HOPO), which served to suppress the racemization. The inner salt methyl-N-(triethylammoniosulfonyl)carbamate is the so-called Burgess reagent, usually used to convert amide groups into nitriles by dehydration. Here, in the last step, the P1′ nitrile group was formed by using Burgess reagent. As a result of the work-up procedure, the product is isolated as a methyl tert-butyl ether (MTBE) solvate. Nirmarelvir was obtained with an overall yield of 49% by the synthetic route shown in SchemeScheme 4 4 [9][16][19][4,23,26].
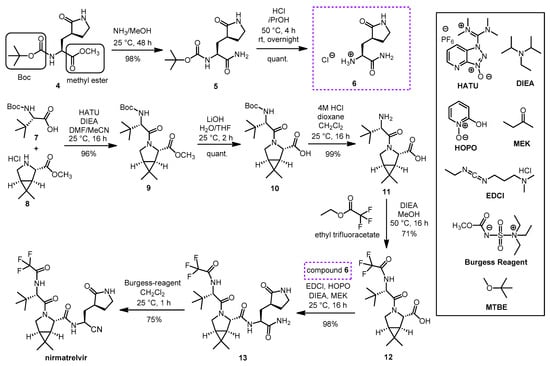
Scheme 3. Synthesis of nirmatrelvir. (HATU: O-(7-azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate, DIEA: diisopropyl-ethylamine, HOPO: 2-hydroxypyridine 1-oxide, MEK: methyl ethyl ketone, EDCl: 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride, DMF: N,N-dimethylformamide, THF: tetrahydrofuran, MTBE: methyl tert-butyl ether.).
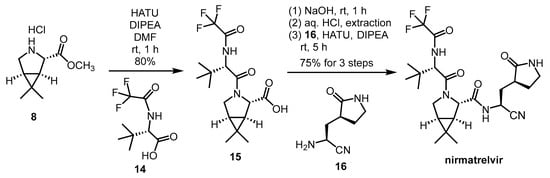
Scheme 4. Alternative synthesis of nirmatrelvir. (HATU: O-(7-azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate, DIPEA = DIEA: diisopropyl-ethylamine).
Of note, compound 8, a bicyclic proline methyl ester derivative containing three chiral centers, is a key building unit in the synthesis of nirmatrelvir. Since 8 is also a building block of boceprevir, many different synthetic routes have been described for it [20][27].
Another synthetic method (Scheme 4) was reported by Zhao et al. This is a much shorter and simpler reaction route, consisting of only two amide couplings and a deprotection step starting from 8, and it also gives free nirmatrelvir instead of MTBE solvate. The overall yield of the presented synthesis steps was 60%, but it should be noted that the synthesis of building blocks 14 and 16 has not been reported [21][28].
In 2023, Ruijter, Turner and co-workers reported a remarkable new synthetic approach to nirmatrelvir based on a highly diastereoselective Ugi-type three-component reaction (Scheme 5) [22][29]. One of the key building blocks, the chiral bicyclic imine 18, was prepared by enantioselective oxidative desymmetrization of meso-pyrrolidine 17 with monoamine oxidase N (MAO-N) [23][30]. Due to the high volatility of 18, it was isolated in the form of its crystalline bisulfite adduct 19 [24][31], from which the free imine 18 was generated in situ for the three-component reaction by basic treatment. The isocyanide building block 23 was prepared from the known Boc-protected amino ester 4. To ensure the appropriate stability and reactivity of isocyanide 23, the C-terminus of 4 was converted to the protected primary alcohol 21 in two steps including reduction (20) and benzoylation. Boc deprotection followed by immediate formylation resulted in formamide 22, from which cyanide 23 was obtained by dehydration with triphosgene. Ugi-type reaction [25][32] of the commercially available carboxylic acid 14 with the in situ prepared imine (18) and isocyanide (23) afforded the nirmatrelvir core 24 in 68% yield and high diastereoselectivity. After debenzoylation, the oxidative conversion of the C-terminal primary alcohol of 25 to a nitrile was performed in a one-pot process, combining PhI(OAc)2/TEMPO-mediated oxidation with ammonium acetate as the nitrogen source. This multicomponent synthesis proceeded in six steps yielding nirmatrelvir with an overall yield of 46%.
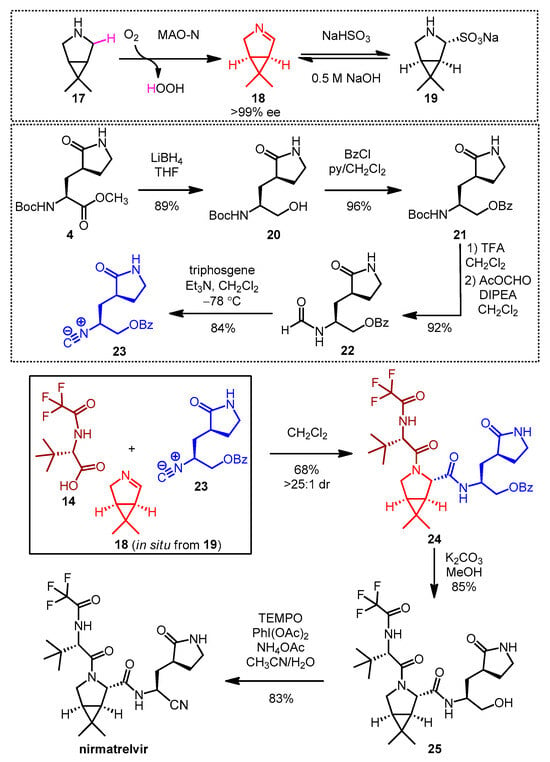
Scheme 5. Multicomponent synthesis of nirmatrelvir. (MAO-N: monoamine oxidase N, ee: enantiomeric excess, THF: tetrahydrofuran, BzCl: benzoyl chloride, py: pyridine, TFA: trifluoroacetic acid, DIPEA: disopropylethylamine, Et3N: triethylamine, dr: diastereomeric ratio, TEMPO: (2,2,6,6-tetramethylpiperidine-1-yl)oxyl, PhI(OAc)2: (diacetoxyiodo)benzene).
2.4. Synthesis and SAR Study of Nirmatrelvir Analogs
Chia and co-workers synthesized a small library of nirmatrelvir analogs with different P1′ moieties (Figure 4) to study the role of the warhead in antiviral activity. The compounds were tested for their enzyme inhibitory activity against the 3CLpro (Mpro) protease of SARS-CoV-2 and hCoV 229E (a human coronavirus that causes the common cold), and their antiviral activity against hCoV 229E. Derivatives without a warhead and with primary alcohol, primary amide, or methyl ester warheads were ineffective.
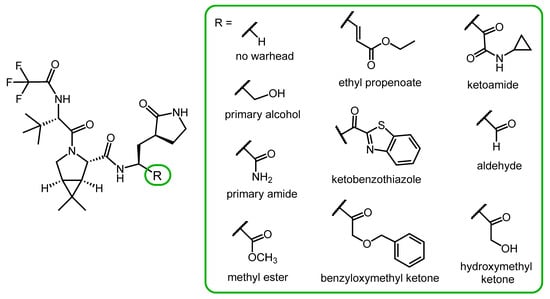
Figure 4.
Nirmatrelvir derivatives with alternative warheads.
Derivatives containing ethyl propenoate, ketobenzothiazole, benzyloxymethyl ketone, ketoamide and aldehyde warheads showed similar or better Mpro inhibitory effect than nirmatrelvir. However, in a cell-based assay, their antiviral activity against hCoV 229E was inferior to nirmatrelvir, probably due to their lower cell penetration ability.
In this compound library, a derivative with a hydroxymethyl ketone warhead was the most potent one, exerting stronger protease inhibitory activity and similar anti-hCoV activity to nirmatrevil. However, a serious limitation of the study is that the cell-based antiviral tests were only performed with the hCoV 229 coronavirus, not with SARS-CoV-2 [26][33].
2.5. Novel Covalent and Non-Covalent Inhibitors of SARS-CoV-2 M
pro
Several other peptidomimetic inhibitors of SARS-CoV-2 Mpro (Figure 5A) have been developed with different warheads, including an epoxide ring (26), a fluoromethyl group (27), a cinnamic ester (28) and a vinyl ester (29). In the latter two compounds, the α,β-unsaturated ester warhead acts as a Michael acceptor, reacting with the thiolate group of MPro Cys145, thus forming an irreversible covalent adduct with the enzyme [12][27][28][6,34,35].
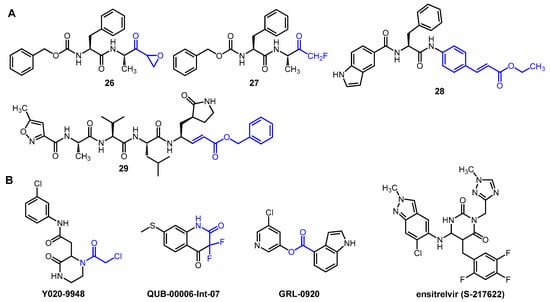
Figure 5. Selected examples of novel peptidic (A) and non-peptidic (B) inhibitors of Mpro. Electrophilic warheads in covalent inhibitors are highlighted in blue.
Interesting non-peptidic inhibitors were also identified (Figure 5B), such as the commercially available piperazine-2 derivative Y020-9948 with an α-chloroacetamide warhead. Compound QUB-00006-Int-07 was developed based on in silico studies. It contains an α,α-difluoroamide group in a benzene-fused six-membered ring. Esters, such as GRL-0920, also showed significant enzyme inhibitory activity due to the electrophilic nature of the ester group, which makes it a suitable warhead [12][6].
Non-covalent inhibitors represent an attractive alternative in the development of anti-coronavirus agents. They do not have an electrophilic warhead, so they only form secondary interactions with the active site, such as H-bonds, hydrophobic stacking, and van der Waals forces. For this reason, they generally show lower reactivity but better selectivity than covalent inhibitors [27][34]. An important representative of non-covalent inhibitors of SARS-CoV-2 Mpro is ensitrelvir (S-217622). This compound fits into the S1 and S2 sub-pockets of the active site, where it forms H-bonding and π-π interactions with the enzyme. Ensitrelvir is approved for COVID-19 in Japan and marketed under the brand name Xocova. Despite its proven efficacy, it is insufficient for hard endpoints such as mortality or hospitalization, which, along with other problems (e.g., the occurrence of resistance), may limit its future use [29][36].