Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Marino Paroli.
Giant cell arteritis (GCA) is a noninfectious granulomatous vasculitis of unknown etiology affecting individuals older than 50 years. Two forms of GCA have been identified: a cranial form involving the medium-caliber temporal artery causing temporal arteritis (TA) and an extracranial form involving the large vessels, mainly the thoracic aorta and its branches. GCA generally affects individuals with a genetic predisposition, but several epigenetic (micro)environmental factors are often critical for the onset of this vasculitis.
- giant cell arteritis
- innate immunity
- adaptive immunity
1. Introduction
Giant cell arteritis (GCA) is a granulomatous vasculitis involving medium- and large-caliber arterial vessels [1,2][1][2]. GCA typically affects individuals older than 50 years, most frequently women, with a peak incidence between the seventh and eighth decades of life [3]. For a long time, it was thought that GCA was a disease involving only the temporal artery, giving rise to temporal arteritis (TA). Subsequently, it was clarified that GCA can also involve large-caliber vessels. It was therefore underlined that GCA should be classified into a cranial form (c-GCA) and an extracranial or large vessel form (LV-GCA). LV-GCA mainly involves the thoracic aorta and its branches and is the main cause of noninfectious aortitis in humans [4].
Epidemiological data on GCA can sometimes appear controversial because they depend on the different designs of the studies conducted [5]. The prevalence of LV-GCA is probably underestimated because of the difficulty in diagnosing this form of vasculitis compared with TA. However, although there are different data depending on the geographical area considered, through a large meta-analysis, an overall incidence of 10 cases and a prevalence of 51.74 cases per 100,000 people was calculated [6]. The highest incidence is in Scandinavian countries, with a significant north–south gradient in European countries [7]. The higher prevalence of GCA in the female sex was confirmed in a 15-year national retrospective study of inpatients in the United States. Specifically, it found that females accounted for 71.9% of patients with GCA, albeit with a lower mortality risk than males [8].
2. Giant Cell Arteritis and Polymyalgia Rheumatica
A distinctive feature of GCA is its close association with polymyalgia rheumatica (PMR). PMR is a musculoskeletal inflammatory disease characterized by an acute onset with typical signs and symptoms, including morning stiffness of the scapular girdle, anemia, fever, fatigue, and weight loss [13][9]. Several evidences indicate that the two conditions are not only often associated, but share similar pathogenetic mechanisms [14][10] and epidemiological characteristics, such as older age of onset, increased incidence and prevalence in northern European countries, and higher prevalence in the female sex [9][11]. PMR is about 2–3 times more common than GCA and is present in about 50% of patients with this vasculitis and can occur before, at the same time, or after the onset of GCA. On the other hand, GCA is diagnosed in 15–20% of PMR patients [15][12]. It is also interesting to note that asymptomatic GCA has been demonstrated by biopsy or ultrasonography [16][13] or detected at the autopsy [17][14] in a significant number of subjects with PMR, suggesting that the link between the two conditions is probably underestimated. The hypothesis that the two diseases may be different stages of the same disease was first formulated in 1972 [18][15]. More recently, based on further experimental evidence, it has been proposed that failure of peripheral tolerance, breakdown of tissue barriers, and granuloma formation constitute successive stages of a single process proceeding from PMR to GCA. This led to the formulation of the combined term GCA-PMR spectrum disease (GPSD) to define a single comprehensive disease entity [19,20][16][17]. An international committee recently recommended that the close temporal and pathogenetic correlation between GCA and PMR should be closely considered in a treat-to-target approach [21][18]. The European Alliance of Rheumatology Associations (EULAR) recently updated its recommendations for the management of GCA in the more general context of large vessel vasculitis [22][19].3. Immunopathogenesis
3.1. The Role of Innate Immune System
3.1.1. The Macrophages and Neutrophiles
Macrophages have been found to infiltrate the arterial wall of patients with GCA. These cells of the innate immune system are one of the main cell types involved in granuloma formation and are recruited into the vessel wall by DCs and T cells [34,50][20][21]. Attracted to the site of inflammation by soluble factors, particularly chemokines, macrophages are, in turn, capable of producing high levels of active substances, including matrix metalloproteinase 9 (MMP9) [51][22]. This enzyme degrades the arterial wall matrix, allowing the macrophages themselves to penetrate deeper into the adventitious layer of the vessel, where they can induce local recruitment of additional proinflammatory cells. M1-type macrophages, characterized by high proinflammatory activity, tend to localize between the media and adventitia, amplifying damage to the vascular wall through the production of cytokines IL-1 and IL-6 and of reactive oxygen species (ROS). Anti-inflammatory M2-type macrophages, on the other hand, tend to localize at the border between the media and vascular intima [50,52,53][21][23][24]. This subgroup of macrophages produces high amounts of VEGF. Endothelial changes induced by VEGF release are thought to be crucially involved in the vascular remodeling observed during GCA, with thickening of the vascular wall [50,54][21][25]. It should be noted that in GCA, macrophages tend to form special multinucleated cells after fusion of activated cells, so-called giant cells, which are the hallmark of this vasculitis. These cells usually result from resistance to degradation of the phagocytosed material [55][26]. Giant cells have been found in about one-third of temporal artery biopsies from patients with c-GCA [56,57,58][27][28][29]. Neutrophils also play an important role in the pathogenesis of GCA. Neutrophil activation after stimulation by danger signals results in the local release of pro-inflammatory cytokines, including IL-6 [59][30] and IL-17A [28,60][31][32]. Neutrophils also produce enzymes capable of disrupting the extracellular matrix and attacking invading pathogens. Activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase produces a respiratory burst through the production of highly reactive oxygen species (ROS) necessary to kill microbes [28,61,62,63][31][33][34][35]. Neutrophils also form the neutrophil extracellular traps (NETs). NETs are composed of filaments of nuclear material capable of trapping and killing various pathogens. The formation of NETs (NETosis) can follow the apoptosis of neutrophils or occur together with the preservation of neutrophil viability. NET formation can be initiated by various stimuli, including microorganism components, cytokines, immune complexes, autoantibodies or platelets. Exposure to such signals induces disassembly of the neutrophil cytoskeleton, chromatin decondensation with citrullination of histones, and formation of bactericidal substances such as myeloperoxidase (MPO) and pro-coagulant factors as well. The process of NETosis depends strictly on ROS production by NAPDH oxidase and/or activation of mitochondria [64][36]. NET formation may be an important source of autoantigen, as demonstrated in ANCA-associated vasculitis [65][37]. Recently, NETs containing pro-inflammatory cytokines were found in the temporal biopsy of GCA patients [28][31].3.1.2. The Dendritic Cells
A crucial role in the pathogenesis of GCA is played by dendritic cells (DCs) that reside in the space between the media and adventitia of the arterial wall [53,66][24][38]. Dendritic cells express toll-like receptors (TLRs) on their surface, which can recognize specific pathogen-associated molecular patterns (PAMPs) derived from components of microorganisms and/or damage-associated molecular patterns (DAMPs) that recognize various proteins, including fragments of necrotic self-cells [67,68][39][40]. Large numbers of DCs have been shown to reside at the level of the vascular wall in patients with GCA [69][41]. It has been shown that such cells can be recruited to this site through the expression of the C-C chemokine receptor 7 (CCR7). This receptor selectively binds its ligands CCL19 and CCL21, which are abundantly present at the level of vascular tissue. CCL2 is also involved in the recruitment of several cells of the innate and adaptative immune system to the artery wall [70,71][42][43]. Once activated, DCs can induce the activation of macrophages with subsequent amplification of the inflammatory cascade [66,72,73][38][44][45]. In addition, DCs could play a key role in the presentation of a putative antigen to T lymphocytes, thus contributing to their activation of cytokine production [74][46] and granuloma formation [75][47]. It has also been suggested that DCs are abnormally activated in patients with GCA due to a deficit in the expression of programmed death ligand 1 (PD-L1), which normally promotes immunosuppression [76,77][48][49]. In this context, PD-L1-deficient myeloid DCs were hypothesized to facilitate the recruitment of CD4+ T cells into the vascular wall in GCA [74][46] while plasmacytoid DCs could activate CD8+ T cells [78][50]. In support of the role of defective PD-L1 expression by DCs in the pathogenesis of GCA, the possible occurrence of this vasculitis was observed during cancer immunotherapy with immune checkpoint inhibitors (ICIs). These agents selectively block the interaction between PD-L1 and the PD-L1 ligand.3.2. The Involvement of Adaptive Immunity
3.2.1. The Role of T Cells
The first clue suggesting the involvement of T cells in GCA was the finding that the immunohistology lesions in the wall of vessels affected by this vasculitis are granulomatous in nature [1]. Granulomas are known to consist of both cells of the innate system, such as macrophages, and cells of the adaptive system, including T cells that aggregate to form well-ordered structures in the course of inflammation. Further characterization of the subtypes of T cells infiltrating the arterial wall revealed the presence of two major subsets of CD4+ T cells, namely Th1 and Th17 cells [75][47]. Although granulomas are generally detected in the course of infections [83][51], an infectious response to a viral/bacterial antigen has never been convincingly demonstrated in the case of GCA, as discussed above in more detail [84][52]. However, early research aimed at characterizing the antigenic specificity of T cells in GCA found that it was restricted to a few antigens [85][53]. Supporting the role of a microbial antigen in shaping the Th1 cell repertoire in GCA, at least as an initial disease trigger, is the finding that the TCR rearrangement of T cells infiltrating the right and left temporal artery was identical in arteries on both sides of the skull in individual patients [86][54]. Th1 and Th17 cells are believed to play different roles in the pathogenesis of GCA. Th1 cells are characterized by the production of IFN-γ, and their differentiation is controlled by IL12/IFN-γ axis [87,88][55][56]. It has been reported that the presence of high levels of IFN-γ in the vascular wall of patients with GCA is strongly associated with the tendency to develop ischemic vascular injury [89][57]. In addition, this cytokine has been found to be elevated years before the onset of GCA [90][58]. It should be noted that, unlike Th17 cells, Th1 cells are not sensitive to steroid therapy. This fact confirms an additional unmet need for GCA therapy and justifies the effort to identify new therapeutic agents that can also be effective on this important subgroup of cells [53,91][24][59]. As for Th17 cells, they have been found in high concentrations in both the peripheral blood and the inflammatory infiltrate of the vascular wall of patients with GCA [75,92,93][47][60][61]. Th17 cells are characterized by the production of IL-17 [94][62], an interleukin that facilitates macrophage recruitment and defense against infections [95,96][63][64]. IL-17 induces the production by target cells of many pro-inflammatory molecules, such as IL-6 and chemokines, including CXCL8 [97][65], thereby amplifying the downstream inflammatory reaction. It has been clearly established that Th17 cells are involved not only in defense against pathogens but also in the induction of immunopathological phenomena and autoimmunity [98,99][66][67]. The characteristic plasticity of Th17 cells in transforming, under particular stimuli from the microenvironment, into cells with different phenotypes, such as Th1 cells or regulatory T cells (Treg) [100][68] makes it further complex to study their role in the immunopathogenesis of GCA and autoimmunity in general. An important role in the pathogenesis of GCA is played by Treg. As mentioned above, these cells are known to be able to differentiate into Th17 cells and vice versa [101][69]. It was clarified that IL-6, together with tissue growth factor-β, promotes polarization toward the Th17 cell phenotype, whereas IL-6 alone induces differentiation of Treg [102][70]. The presence of IL-23 in the microenvironment produced by cells of the innate system further inhibits the expression of the transcription factor forkhead box P3 (FOXP3) necessary for Treg differentiation. It facilitates the maintenance of the Th17 phenotype and its survival [103,104][71][72]. It has been shown that Treg present at the level of the vessel wall in GCA are unable to perform their regulatory function. Therefore, these cells participate in the pathogenic process instead of inhibiting the pro-inflammatory activity of T-cell subpopulations. Interestingly, therapeutic blockade of IL-6R is able to inhibit these pathogenic Treg in CGA, restoring their anti-inflammatory function, whereas this is not the case with GC therapy [105][73]. It should be further emphasized that the differentiation of T cell precursors into the various T cell subpopulations depends critically on soluble factors produced by cells of the innate immune system, and in particular, DCs [106][74]. DCs can thus be considered the conductors of the T-cell response orchestra. As already discussed, the differentiation of Th0 cells into Th1 cells depends mainly on the presence of IL-12, while the differentiation and stabilization of Th17 cells depend on the presence of IL-23 [107][75]. It has been hypothesized that in GCA, as yet unknown stimuli capable of activating different TLRs on DCs may determine the location and severity of this vasculitis through the induction of responses by specific subpopulations of T cells [66,69,108][38][41][76]. In this regard, studies in animal models have shown that TLR4 stimulation on dendritic cells induces transmural arterial inflammation, whereas TLR5 activation is rather related to perivascular inflammation [72][44]. Other subpopulations of CD4+ T cells that have been described to play an important role in GCA are Th2 and Th9 cells, which produce IL-33 and IL-9, respectively [109,110][77][78]. Further studies are needed to clarify the role of these CD4+ T-cell subsets in the pathogenesis of GCA. CD8+ T cells have been less studied than CD4+ T cells in terms of their pathogenetic role in GCA. Although it has been reported that these cells may not play a significant role in the inflammatory process of GCA due to their anergy related to the advanced age of GCA patients [111,112,113][79][80][81], on the contrary, many studies support the central role of both cytotoxic and regulatory CD8+ T cells in the pathogenesis of this vasculitis. CD8+ T cells with increased ability to produce granzyme A have been reported to be present in the peripheral blood of patients with GCA, and their numbers decreased after GC therapy [114][82]. Temporal artery tissue has also been shown to express natural killer group 2D (NKG2D) ligand that can stimulate the cytotoxic activity of NKG2D receptor-expressing CD8+ T cells [115][83]. As for CD8+ regulatory cells (CD8+ Treg), these are dysfunctional in GCA, like their CD4+ counterpart [116][84]. The reduced anti-inflammatory activity of this anti-inflammatory CD8+ T cell subset has been attributed to NADPH oxidase 2 deficiency [117][85]. Finally, it should be noted that memory T cells residing in the vascular wall of patients with GCA have been shown to contribute to the renewal and persistent presence of proinflammatory T cells at the vascular inflammatory site [118,119,120][86][87][88].3.2.2. The Role of B Cells
Initial studies had suggested a possible role of autoantibodies produced by B cells in the pathogenesis of GCA. These findings, however, have not subsequently been confirmed [121,122][89][90]. In more recent studies, B cells and plasma cells have been identified in high numbers in the temporal artery of patients with this form of GCA [123][91]. In this case, these cells were found to form together with T lymphocytes, dendritic cells, and high endothelial venules (HEVs). These follicle-like structures have been termed tertiary arterial lymphoid organs (ATLOs) [124][92]. ATLOs are predominantly located near granulomas, suggesting that communication between the two structures can occur. Such proximity may, therefore, facilitate macrophage activation in granulomas through the arrival of cytokines and other soluble factors produced by cellular interactions within ATLOs. In further support of the key role of B cells in the pathogenesis of GCA, the B-cell-activating growth factor (BAFF) and the proliferation-induced ligand APRIL, both produced by endothelial cells and VSMCs, have been found in high concentrations at the tissue level in patients with GCA [124][92]. In a recent study, it was shown that, in the active phase of the disease, high numbers of B cells can be identified in the arterial wall of large vessels of GCA patients while decreasing in peripheral blood, demonstrating tissue-selective recruitment of these lymphocytes [125,126][93][94]. The potential role of B cells in the pathogenesis of GCA is also suggested by the description of two cases that significantly improved following therapy with the anti-CD20 antibody rituximab that depletes B cells [127,128][95][96]. The absence of specific antibodies during GCA suggests that these cells play a different role in the immune response rather than contributing to humoral immunity, including the production of soluble factors or antigen presentation to T lymphocytes [129,130,131][97][98][99]. The role of B cells in the pathogenesis of GCA is further underscored by the observation that during the acute stages of the disease, their numbers in the peripheral blood tend to decrease. This denotes their redistribution to the vascular tissue site of inflammation [125,132][93][100]. Figure 1 summarizes the cellular mechanisms involved in the pathogenesis of GCA.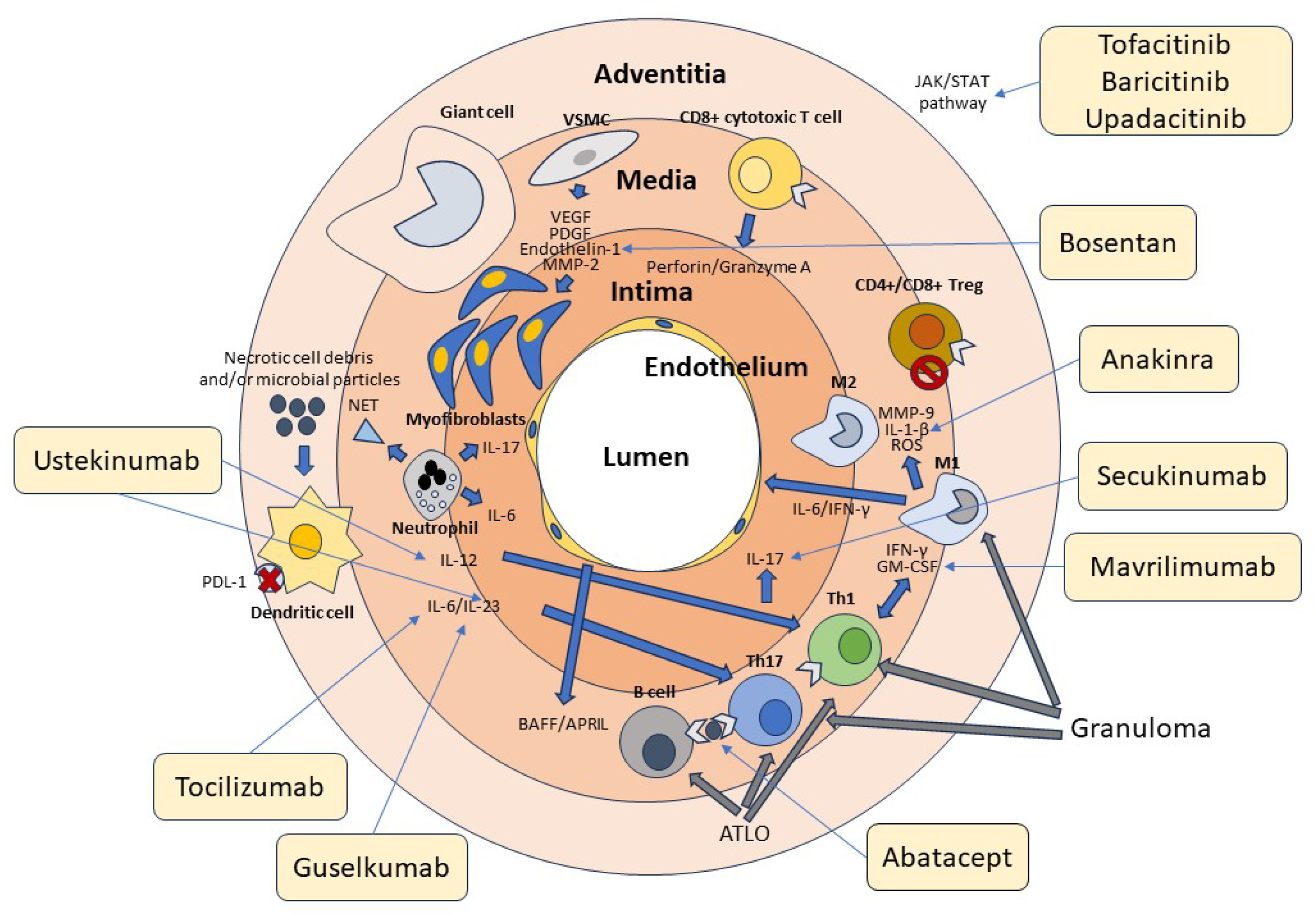
Figure 1. Cellular and molecular pathogenesis of GCA. Cells of the innate and adaptive immune system and the vascular wall contribute to the pathogenesis of GCA. Adventitious dendritic cells are activated by unknown molecules of microbial and/or cellular origin. Dendritic cells in GCA are typically defective in the expression of the immunosuppressive surface molecule PD-L1, as indicated in the figure by the red cross. Dendritic cells produce IL-12, which promotes the differentiation of Th1 cells, and Il-6 and IL-23, which contribute to the differentiation and stabilization of the phenotype of Th17 cells. T lymphocytes are activated by both dendritic cells and B lymphocytes through the presentation of a putative antigen. Th1 cells produce IFN-γ and GM-CSF, while Th17 cells produce IL-17. These cytokines activate M1 and M1 macrophages, which in turn produce MMP-9, IL1-β, and ROS, contributing to media destruction. Some macrophages, unable to kill the phagocytosed material, transform into giant cells. CD4+ and CD8+ Treg cells participate in the inflammatory reaction, being deficient in their immunosuppressive function, as indicated by the stop symbol. CD8+ cytotoxic T cells and neutrophils producing proinflammatory cytokines and NETs play an additional role in the pathogenesis of GCA. At the vascular level, damaged VSMCs produce VEGF, PDGF, endothelin-1, and MMP-2, which promote their differentiation into myofibroblasts. These cells cause thickening of the intima and subsequent vascular stenosis. Many cytokines recognize JAK-associated receptors and amplify the inflammatory cascade through the activation of the JAK/STAT signaling pathway. T and B cells aggregate to form tertiary follicular structures (ATLO), whereas T cells and macrophages are the main components of granulomas. The light-yellow boxes show the main drugs available to date that inhibit the various factors involved in the pathogenesis of GCA, as indicated by the thin arrows. Of these, only tocilizumab has been officially approved for the treatment of this vasculitis.
References
- Weyand, C.M.; Goronzy, J.J. Medium- and large-vessel vasculitis. N. Engl. J. Med. 2003, 349, 160–169.
- Espitia, O.; Samson, M.; Le Gallou, T.; Connault, J.; Landron, C.; Lavigne, C.; Belizna, C.; Magnant, J.; de Moreuil, C.; Roblot, P.; et al. Comparison of idiopathic (isolated) aortitis and giant cell arteritis-related aortitis. A French retrospective multicenter study of 117 patients. Autoimmun. Rev. 2016, 15, 571–576.
- Salvarani, C.; Cantini, F.; Hunder, G.G. Polymyalgia rheumatica and giant-cell arteritis. Lancet 2008, 372, 234–245.
- Carvajal Alegria, G.; van Sleen, Y.; Graver, J.C.; Sandovici, M.; Devauchelle-Pensec, V.; Brouwer, E.; Cornec, D. Aortic involvement in giant cell arteritis. Jt. Bone Spine 2021, 88, 105045.
- Watts, R.A.; Hatemi, G.; Burns, J.C.; Mohammad, A.J. Global epidemiology of vasculitis. Nat. Rev. Rheumatol. 2022, 18, 22–34.
- Li, K.J.; Semenov, D.; Turk, M.; Pope, J. A meta-analysis of the epidemiology of giant cell arteritis across time and space. Arthritis Res. Ther. 2021, 23, 82.
- Sharma, A.; Mohammad, A.J.; Turesson, C. Incidence and prevalence of giant cell arteritis and polymyalgia rheumatica: A systematic literature review. Semin. Arthritis Rheum. 2020, 50, 1040–1048.
- Idowu, A.B.; Khandwala, P.; Tan, I.J. Race and Gender on the Mortality of Giant Cell Arteritis in Hospitalized Patients: A 15-Year National Inpatient Study. Cureus 2023, 15, e46165.
- Raheel, S.; Shbeeb, I.; Crowson, C.S.; Matteson, E.L. Epidemiology of Polymyalgia Rheumatica 2000–2014 and Examination of Incidence and Survival Trends Over 45 Years: A Population-Based Study. Arthritis Care Res. 2017, 69, 1282–1285.
- Dejaco, C.; Duftner, C.; Buttgereit, F.; Matteson, E.L.; Dasgupta, B. The spectrum of giant cell arteritis and polymyalgia rheumatica: Revisiting the concept of the disease. Rheumatology 2017, 56, 506–515.
- Buttgereit, F.; Matteson, E.L.; Dejaco, C. Polymyalgia Rheumatica and Giant Cell Arteritis. JAMA 2020, 324, 993–994.
- Dejaco, C.; Brouwer, E.; Mason, J.C.; Buttgereit, F.; Matteson, E.L.; Dasgupta, B. Giant cell arteritis and polymyalgia rheumatica: Current challenges and opportunities. Nat. Rev. Rheumatol. 2017, 13, 578–592.
- Schmidt, W.A.; Gromnica-Ihle, E. Incidence of temporal arteritis in patients with polymyalgia rheumatica: A prospective study using colour Doppler ultrasonography of the temporal arteries. Rheumatology 2002, 41, 46–52.
- Hamrin, B.; Jonsson, N.; Hellsten, S. “Polymyalgia arteritica”. Further clinical and histopathological studies with a report of six autopsy cases. Ann. Rheum. Dis. 1968, 27, 397–405.
- Fauchald, P.; Rygvold, O.; Oystese, B. Temporal arteritis and polymyalgia rheumatica. Clinical and biopsy findings. Ann. Intern. Med. 1972, 77, 845–852.
- Salvarani, C.; Cantini, F.; Boiardi, L.; Hunder, G.G. Polymyalgia rheumatica and giant-cell arteritis. N. Engl. J. Med. 2002, 347, 261–271.
- Tomelleri, A.; van der Geest, K.S.M.; Khurshid, M.A.; Sebastian, A.; Coath, F.; Robbins, D.; Pierscionek, B.; Dejaco, C.; Matteson, E.; van Sleen, Y.; et al. Disease stratification in GCA and PMR: State of the art and future perspectives. Nat. Rev. Rheumatol. 2023, 19, 446–459.
- Dejaco, C.; Kerschbaumer, A.; Aletaha, D.; Bond, M.; Hysa, E.; Camellino, D.; Ehlers, L.; Abril, A.; Appenzeller, S.; Cid, M.C.; et al. Treat-to-target recommendations in giant cell arteritis and polymyalgia rheumatica. Ann. Rheum. Dis. 2023, 83, 48–57.
- Hellmich, B.; Agueda, A.; Monti, S.; Buttgereit, F.; de Boysson, H.; Brouwer, E.; Cassie, R.; Cid, M.C.; Dasgupta, B.; Dejaco, C.; et al. 2018 Update of the EULAR recommendations for the management of large vessel vasculitis. Ann. Rheum. Dis. 2020, 79, 19–30.
- Corbera-Bellalta, M.; Planas-Rigol, E.; Lozano, E.; Terrades-Garcia, N.; Alba, M.A.; Prieto-Gonzalez, S.; Garcia-Martinez, A.; Albero, R.; Enjuanes, A.; Espigol-Frigole, G.; et al. Blocking interferon gamma reduces expression of chemokines CXCL9, CXCL10 and CXCL11 and decreases macrophage infiltration in ex vivo cultured arteries from patients with giant cell arteritis. Ann. Rheum. Dis. 2016, 75, 1177–1186.
- Koster, M.J.; Warrington, K.J. Giant cell arteritis: Pathogenic mechanisms and new potential therapeutic targets. BMC Rheumatol. 2017, 1, 2.
- Watanabe, R.; Maeda, T.; Zhang, H.; Berry, G.J.; Zeisbrich, M.; Brockett, R.; Greenstein, A.E.; Tian, L.; Goronzy, J.J.; Weyand, C.M. MMP (Matrix Metalloprotease)-9-Producing Monocytes Enable T Cells to Invade the Vessel Wall and Cause Vasculitis. Circ. Res. 2018, 123, 700–715.
- Wagner, A.D.; Goronzy, J.J.; Weyand, C.M. Functional profile of tissue-infiltrating and circulating CD68+ cells in giant cell arteritis. Evidence for two components of the disease. J. Clin. Investig. 1994, 94, 1134–1140.
- Weyand, C.M.; Goronzy, J.J. Immune mechanisms in medium and large-vessel vasculitis. Nat. Rev. Rheumatol. 2013, 9, 731–740.
- Weyand, C.M.; Liao, Y.J.; Goronzy, J.J. The immunopathology of giant cell arteritis: Diagnostic and therapeutic implications. J. Neuroophthalmol. 2012, 32, 259–265.
- Anderson, J.M. Multinucleated giant cells. Curr. Opin. Hematol. 2000, 7, 40–47.
- Cavazza, A.; Muratore, F.; Boiardi, L.; Restuccia, G.; Pipitone, N.; Pazzola, G.; Tagliavini, E.; Ragazzi, M.; Rossi, G.; Salvarani, C. Inflamed temporal artery: Histologic findings in 354 biopsies, with clinical correlations. Am. J. Surg. Pathol. 2014, 38, 1360–1370.
- Armstrong, A.T.; Tyler, W.B.; Wood, G.C.; Harrington, T.M. Clinical importance of the presence of giant cells in temporal arteritis. J. Clin. Pathol. 2008, 61, 669–671.
- Chatelain, D.; Duhaut, P.; Schmidt, J.; Loire, R.; Bosshard, S.; Guernou, M.; Pellet, H.; Piette, J.C.; Sevestre, H.; Ducroix, J.P.; et al. Pathological features of temporal arteries in patients with giant cell arteritis presenting with permanent visual loss. Ann. Rheum. Dis. 2009, 68, 84–88.
- Bank, U.; Reinhold, D.; Kunz, D.; Schulz, H.U.; Schneemilch, C.; Brandt, W.; Ansorge, S. Effects of interleukin-6 (IL-6) and transforming growth factor-beta (TGF-beta) on neutrophil elastase release. Inflammation 1995, 19, 83–99.
- Palamidas, D.A.; Argyropoulou, O.D.; Georgantzoglou, N.; Karatza, E.; Xingi, E.; Kapsogeorgou, E.K.; Anagnostopoulos, C.D.; Lazaris, A.C.; Ritis, K.; Goules, A.V.; et al. Neutrophil extracellular traps in giant cell arteritis biopsies: Presentation, localization and co-expression with inflammatory cytokines. Rheumatology 2022, 61, 1639–1644.
- Taylor, P.R.; Roy, S.; Leal, S.M., Jr.; Sun, Y.; Howell, S.J.; Cobb, B.A.; Li, X.; Pearlman, E. Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORgammat and dectin-2. Nat. Immunol. 2014, 15, 143–151.
- Wang, L.; Ai, Z.; Khoyratty, T.; Zec, K.; Eames, H.L.; van Grinsven, E.; Hudak, A.; Morris, S.; Ahern, D.; Monaco, C.; et al. ROS-producing immature neutrophils in giant cell arteritis are linked to vascular pathologies. JCI Insight 2020, 5, e139163.
- Nadkarni, S.; Dalli, J.; Hollywood, J.; Mason, J.C.; Dasgupta, B.; Perretti, M. Investigational analysis reveals a potential role for neutrophils in giant-cell arteritis disease progression. Circ. Res. 2014, 114, 242–248.
- Foell, D.; Hernandez-Rodriguez, J.; Sanchez, M.; Vogl, T.; Cid, M.C.; Roth, J. Early recruitment of phagocytes contributes to the vascular inflammation of giant cell arteritis. J. Pathol. 2004, 204, 311–316.
- Wigerblad, G.; Kaplan, M.J. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat. Rev. Immunol. 2023, 23, 274–288.
- Paroli, M.; Gioia, C.; Accapezzato, D. New Insights into Pathogenesis and Treatment of ANCA-Associated Vasculitis: Autoantibodies and Beyond. Antibodies 2023, 12, 25.
- Ma-Krupa, W.; Jeon, M.S.; Spoerl, S.; Tedder, T.F.; Goronzy, J.J.; Weyand, C.M. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J. Exp. Med. 2004, 199, 173–183.
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384.
- O’Neill, L.; Molloy, E.S. The role of toll like receptors in giant cell arteritis. Rheumatology 2016, 55, 1921–1931.
- Krupa, W.M.; Dewan, M.; Jeon, M.S.; Kurtin, P.J.; Younge, B.R.; Goronzy, J.J.; Weyand, C.M. Trapping of misdirected dendritic cells in the granulomatous lesions of giant cell arteritis. Am. J. Pathol. 2002, 161, 1815–1823.
- Dress, R.J.; Wong, A.Y.; Ginhoux, F. Homeostatic control of dendritic cell numbers and differentiation. Immunol. Cell Biol. 2018, 96, 463–476.
- Corbera-Bellalta, M.; Garcia-Martinez, A.; Lozano, E.; Planas-Rigol, E.; Tavera-Bahillo, I.; Alba, M.A.; Prieto-Gonzalez, S.; Butjosa, M.; Espigol-Frigole, G.; Hernandez-Rodriguez, J.; et al. Changes in biomarkers after therapeutic intervention in temporal arteries cultured in Matrigel: A new model for preclinical studies in giant-cell arteritis. Ann. Rheum. Dis. 2014, 73, 616–623.
- Deng, J.; Ma-Krupa, W.; Gewirtz, A.T.; Younge, B.R.; Goronzy, J.J.; Weyand, C.M. Toll-like receptors 4 and 5 induce distinct types of vasculitis. Circ. Res. 2009, 104, 488–495.
- Song, G.G.; Choi, S.J.; Ji, J.D.; Lee, Y.H. Toll-like receptor polymorphisms and vasculitis susceptibility: Meta-analysis and systematic review. Mol. Biol. Rep. 2013, 40, 1315–1323.
- Samson, M.; Corbera-Bellalta, M.; Audia, S.; Planas-Rigol, E.; Martin, L.; Cid, M.C.; Bonnotte, B. Recent advances in our understanding of giant cell arteritis pathogenesis. Autoimmun. Rev. 2017, 16, 833–844.
- Deng, J.; Younge, B.R.; Olshen, R.A.; Goronzy, J.J.; Weyand, C.M. Th17 and Th1 T-cell responses in giant cell arteritis. Circulation 2010, 121, 906–915.
- Watanabe, R.; Zhang, H.; Berry, G.; Goronzy, J.J.; Weyand, C.M. Immune checkpoint dysfunction in large and medium vessel vasculitis. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H1052–H1059.
- Hid Cadena, R.; Abdulahad, W.H.; Hospers, G.A.P.; Wind, T.T.; Boots, A.M.H.; Heeringa, P.; Brouwer, E. Checks and Balances in Autoimmune Vasculitis. Front. Immunol. 2018, 9, 315.
- Reizis, B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 2019, 50, 37–50.
- Saunders, B.M.; Cooper, A.M. Restraining mycobacteria: Role of granulomas in mycobacterial infections. Immunol. Cell Biol. 2000, 78, 334–341.
- Duhaut, P.; Bosshard, S.; Ducroix, J.P. Is giant cell arteritis an infectious disease? Biological and epidemiological evidence. Presse Med. 2004, 33, 1403–1408.
- Weyand, C.M.; Goronzy, J.J. Giant cell arteritis as an antigen-driven disease. Rheum. Dis. Clin. N. Am. 1995, 21, 1027–1039.
- Weyand, C.M.; Schonberger, J.; Oppitz, U.; Hunder, N.N.; Hicok, K.C.; Goronzy, J.J. Distinct vascular lesions in giant cell arteritis share identical T cell clonotypes. J. Exp. Med. 1994, 179, 951–960.
- Weyand, C.M.; Hicok, K.C.; Hunder, G.G.; Goronzy, J.J. Tissue cytokine patterns in patients with polymyalgia rheumatica and giant cell arteritis. Ann. Intern. Med. 1994, 121, 484–491.
- Weyand, C.M.; Tetzlaff, N.; Bjornsson, J.; Brack, A.; Younge, B.; Goronzy, J.J. Disease patterns and tissue cytokine profiles in giant cell arteritis. Arthritis Rheum. 1997, 40, 19–26.
- Brack, A.; Martinez-Taboada, V.; Stanson, A.; Goronzy, J.J.; Weyand, C.M. Disease pattern in cranial and large-vessel giant cell arteritis. Arthritis Rheum. 1999, 42, 311–317.
- Wadstrom, K.; Jacobsson, L.T.H.; Mohammad, A.J.; Warrington, K.J.; Matteson, E.L.; Jakobsson, M.E.; Turesson, C. Analyses of plasma inflammatory proteins reveal biomarkers predictive of subsequent development of giant cell arteritis: A prospective study. Rheumatology 2023, 62, 2304–2311.
- Weyand, C.M.; Younge, B.R.; Goronzy, J.J. IFN-gamma and IL-17: The two faces of T-cell pathology in giant cell arteritis. Curr. Opin. Rheumatol. 2011, 23, 43–49.
- Samson, M.; Audia, S.; Fraszczak, J.; Trad, M.; Ornetti, P.; Lakomy, D.; Ciudad, M.; Leguy, V.; Berthier, S.; Vinit, J.; et al. Th1 and Th17 lymphocytes expressing CD161 are implicated in giant cell arteritis and polymyalgia rheumatica pathogenesis. Arthritis Rheum. 2012, 64, 3788–3798.
- Terrier, B.; Geri, G.; Chaara, W.; Allenbach, Y.; Rosenzwajg, M.; Costedoat-Chalumeau, N.; Fouret, P.; Musset, L.; Benveniste, O.; Six, A.; et al. Interleukin-21 modulates Th1 and Th17 responses in giant cell arteritis. Arthritis Rheum. 2012, 64, 2001–2011.
- Littman, D.R.; Rudensky, A.Y. Th17 and regulatory T cells in mediating and restraining inflammation. Cell 2010, 140, 845–858.
- Maddur, M.S.; Miossec, P.; Kaveri, S.V.; Bayry, J. Th17 cells: Biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am. J. Pathol. 2012, 181, 8–18.
- Peck, A.; Mellins, E.D. Precarious balance: Th17 cells in host defense. Infect. Immun. 2010, 78, 32–38.
- Matsuzaki, G.; Umemura, M. Interleukin-17 as an effector molecule of innate and acquired immunity against infections. Microbiol. Immunol. 2007, 51, 1139–1147.
- Paroli, M.; Caccavale, R.; Fiorillo, M.T.; Spadea, L.; Gumina, S.; Candela, V.; Paroli, M.P. The Double Game Played by Th17 Cells in Infection: Host Defense and Immunopathology. Pathogens 2022, 11, 1547.
- Sallusto, F.; Lanzavecchia, A. Human Th17 cells in infection and autoimmunity. Microbes Infect. 2009, 11, 620–624.
- Lee, Y.K.; Mukasa, R.; Hatton, R.D.; Weaver, C.T. Developmental plasticity of Th17 and Treg cells. Curr. Opin. Immunol. 2009, 21, 274–280.
- Kleinewietfeld, M.; Hafler, D.A. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin. Immunol. 2013, 25, 305–312.
- Lee, G.R. The Balance of Th17 versus Treg Cells in Autoimmunity. Int. J. Mol. Sci. 2018, 19, 730.
- Kimura, A.; Kishimoto, T. IL-6: Regulator of Treg/Th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835.
- Barbi, J.; Pardoll, D.; Pan, F. Metabolic control of the Treg/Th17 axis. Immunol. Rev. 2013, 252, 52–77.
- Miyabe, C.; Miyabe, Y.; Strle, K.; Kim, N.D.; Stone, J.H.; Luster, A.D.; Unizony, S. An expanded population of pathogenic regulatory T cells in giant cell arteritis is abrogated by IL-6 blockade therapy. Ann. Rheum. Dis. 2017, 76, 898–905.
- Kleinschek, M.A.; Boniface, K.; Sadekova, S.; Grein, J.; Murphy, E.E.; Turner, S.P.; Raskin, L.; Desai, B.; Faubion, W.A.; de Waal Malefyt, R.; et al. Circulating and gut-resident human Th17 cells express CD161 and promote intestinal inflammation. J. Exp. Med. 2009, 206, 525–534.
- Annunziato, F.; Romagnani, S. Do studies in humans better depict Th17 cells? Blood 2009, 114, 2213–2219.
- Han, J.W.; Shimada, K.; Ma-Krupa, W.; Johnson, T.L.; Nerem, R.M.; Goronzy, J.J.; Weyand, C.M. Vessel wall-embedded dendritic cells induce T-cell autoreactivity and initiate vascular inflammation. Circ. Res. 2008, 102, 546–553.
- Ciccia, F.; Alessandro, R.; Rizzo, A.; Raimondo, S.; Giardina, A.; Raiata, F.; Boiardi, L.; Cavazza, A.; Guggino, G.; De Leo, G.; et al. IL-33 is overexpressed in the inflamed arteries of patients with giant cell arteritis. Ann. Rheum. Dis. 2013, 72, 258–264.
- Ciccia, F.; Rizzo, A.; Guggino, G.; Cavazza, A.; Alessandro, R.; Maugeri, R.; Cannizzaro, A.; Boiardi, L.; Iacopino, D.G.; Salvarani, C.; et al. Difference in the expression of IL-9 and IL-17 correlates with different histological pattern of vascular wall injury in giant cell arteritis. Rheumatology 2015, 54, 1596–1604.
- Moskowitz, D.M.; Zhang, D.W.; Hu, B.; Le Saux, S.; Yanes, R.E.; Ye, Z.; Buenrostro, J.D.; Weyand, C.M.; Greenleaf, W.J.; Goronzy, J.J. Epigenomics of human CD8 T cell differentiation and aging. Sci. Immunol. 2017, 2, eaag0192.
- Czesnikiewicz-Guzik, M.; Lee, W.W.; Cui, D.; Hiruma, Y.; Lamar, D.L.; Yang, Z.Z.; Ouslander, J.G.; Weyand, C.M.; Goronzy, J.J. T cell subset-specific susceptibility to aging. Clin. Immunol. 2008, 127, 107–118.
- Reitsema, R.D.; Boots, A.M.H.; van der Geest, K.S.M.; Sandovici, M.; Heeringa, P.; Brouwer, E. CD8+ T Cells in GCA and GPA: Bystanders or Active Contributors? Front. Immunol. 2021, 12, 654109.
- Samson, M.; Ly, K.H.; Tournier, B.; Janikashvili, N.; Trad, M.; Ciudad, M.; Gautheron, A.; Devilliers, H.; Quipourt, V.; Maurier, F.; et al. Involvement and prognosis value of CD8(+) T cells in giant cell arteritis. J. Autoimmun. 2016, 72, 73–83.
- Dejaco, C.; Duftner, C.; Al-Massad, J.; Wagner, A.D.; Park, J.K.; Fessler, J.; Aigelsreiter, A.; Hafner, F.; Vega, S.; Sterlacci, W.; et al. NKG2D stimulated T-cell autoreactivity in giant cell arteritis and polymyalgia rheumatica. Ann. Rheum. Dis. 2013, 72, 1852–1859.
- Collison, J. Vasculitis syndromes: Dysfunctional CD8 TREG cells implicated in GCA. Nat. Rev. Rheumatol. 2016, 12, 314.
- Wen, Z.; Shimojima, Y.; Shirai, T.; Li, Y.; Ju, J.; Yang, Z.; Tian, L.; Goronzy, J.J.; Weyand, C.M. NADPH oxidase deficiency underlies dysfunction of aged CD8+ Tregs. J. Clin. Investig. 2016, 126, 1953–1967.
- Park, C.O.; Kupper, T.S. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat. Med. 2015, 21, 688–697.
- Zhang, H.; Watanabe, R.; Berry, G.J.; Tian, L.; Goronzy, J.J.; Weyand, C.M. Inhibition of JAK-STAT Signaling Suppresses Pathogenic Immune Responses in Medium and Large Vessel Vasculitis. Circulation 2018, 137, 1934–1948.
- Akiyama, M.; Ohtsuki, S.; Berry, G.J.; Liang, D.H.; Goronzy, J.J.; Weyand, C.M. Innate and Adaptive Immunity in Giant Cell Arteritis. Front. Immunol. 2020, 11, 621098.
- Espinoza, L.R.; Jara, L.J.; Silveira, L.H.; Martinez-Osuna, P.; Zwolinska, J.B.; Kneer, C.; Aguilar, J.L. Anticardiolipin antibodies in polymyalgia rheumatica-giant cell arteritis: Association with severe vascular complications. Am. J. Med. 1991, 90, 474–478.
- Salvarani, C.; Baricchi, R.; Macchioni, P.; Carbognani, R.; Morelli, A.; Lodi, L.; Portioli, I. Anticardiolipin antibodies in northern Italian population with PMR/GCA. Am. J. Med. 1992, 92, 712–714.
- Maleszewski, J.J.; Younge, B.R.; Fritzlen, J.T.; Hunder, G.G.; Goronzy, J.J.; Warrington, K.J.; Weyand, C.M. Clinical and pathological evolution of giant cell arteritis: A prospective study of follow-up temporal artery biopsies in 40 treated patients. Mod. Pathol. 2017, 30, 788–796.
- Ciccia, F.; Rizzo, A.; Ferrante, A.; Guggino, G.; Croci, S.; Cavazza, A.; Salvarani, C.; Triolo, G. New insights into the pathogenesis of giant cell arteritis. Autoimmun. Rev. 2017, 16, 675–683.
- van der Geest, K.S.; Abdulahad, W.H.; Chalan, P.; Rutgers, A.; Horst, G.; Huitema, M.G.; Roffel, M.P.; Roozendaal, C.; Kluin, P.M.; Bos, N.A.; et al. Disturbed B cell homeostasis in newly diagnosed giant cell arteritis and polymyalgia rheumatica. Arthritis Rheumatol. 2014, 66, 1927–1938.
- Graver, J.C.; Boots, A.M.H.; Haacke, E.A.; Diepstra, A.; Brouwer, E.; Sandovici, M. Massive B-Cell Infiltration and Organization into Artery Tertiary Lymphoid Organs in the Aorta of Large Vessel Giant Cell Arteritis. Front. Immunol. 2019, 10, 83.
- Bhatia, A.; Ell, P.J.; Edwards, J.C. Anti-CD20 monoclonal antibody (rituximab) as an adjunct in the treatment of giant cell arteritis. Ann. Rheum. Dis. 2005, 64, 1099–1100.
- Mayrbaeurl, B.; Hinterreiter, M.; Burgstaller, S.; Windpessl, M.; Thaler, J. The first case of a patient with neutropenia and giant-cell arteritis treated with rituximab. Clin. Rheumatol. 2007, 26, 1597–1598.
- Baerlecken, N.T.; Linnemann, A.; Gross, W.L.; Moosig, F.; Vazquez-Rodriguez, T.R.; Gonzalez-Gay, M.A.; Martin, J.; Kotter, I.; Henes, J.C.; Melchers, I.; et al. Association of ferritin autoantibodies with giant cell arteritis/polymyalgia rheumatica. Ann. Rheum. Dis. 2012, 71, 943–947.
- Regent, A.; Ly, K.H.; Blet, A.; Agard, C.; Puechal, X.; Tamas, N.; Le-Jeunne, C.; Vidal, E.; Guillevin, L.; Mouthon, L. Contribution of antiferritin antibodies to diagnosis of giant cell arteritis. Ann. Rheum. Dis. 2013, 72, 1269–1270.
- Kistner, A.; Bigler, M.B.; Glatz, K.; Egli, S.B.; Baldin, F.S.; Marquardsen, F.A.; Mehling, M.; Rentsch, K.M.; Staub, D.; Aschwanden, M.; et al. Characteristics of autoantibodies targeting 14-3-3 proteins and their association with clinical features in newly diagnosed giant cell arteritis. Rheumatology 2017, 56, 829–834.
- Carvajal Alegria, G.; Devauchelle-Pensec, V.; Renaudineau, Y.; Saraux, A.; Pers, J.O.; Cornec, D. Correction of abnormal B-cell subset distribution by interleukin-6 receptor blockade in polymyalgia rheumatica. Rheumatology 2017, 56, 1401–1406.
More