Within the concept of continuous spike–waves during slow sleep (CSWS), researchers can find several childhood-onset heterogeneous conditions that share electroencephalograms (EEGs) characterized by a high frequency of paroxysmal abnormalities during sleep, which have negative effects on the cognitive development and behavior of the child. These negative effects may have the characteristics of a clear regression or of a slowdown in development. Seizures are very often present, but not constantly. The above makes it clear why CSWS have been included in epileptic encephalopathies, in which, by definition, frequent EEG paroxysmal abnormalities have an unfavorable impact on cognitive functions, including socio-communicative skills, causing autistic features
[1][2][3][4][5][6][1,2,3,4,5,6], even regardless of the presence of clinically overt seizures. This unfavorable impact has been confirmed in brain functional imaging studies
[7]. In other words, in epileptic encephalopathies, epileptic activity itself (evidenced by EEG paroxysmal abnormalities) contributes to the development of serious cognitive and behavioral disorders, regardless of what might be expected due to the underlying pathology (if any) alone
[8]. However, there have also been authors who aimed at diminishing the importance of CSWS in the development of cognitive deficits
[9]. The first pioneering report that pointed out the connection between continuous EEG paroxysmal abnormalities (spike and wave complexes) and the presence of cognitive disorders should be attributed to Kennedy and Hill, who in 1942 described a child affected by “dementia dysrhythmica infantum”
[10]. Between the 1970s and 1980s, Tassinari et al. described, in a very detailed and modern way, the electroclinical entity characterized by an EEG with diffuse spikes and waves lasting for at least 85% of slow sleep, clinically associated with cognitive decline, seizures, and motor impairment
[11][12][11,12]. This is the typical form of the “electrical status epilepticus during slow-wave sleep” (ESES) that has been included among the epileptic encephalopathies according to the International Classification of the Epilepsies due to marked EEG abnormalities, leading to a cognitive impairment that may cause a severe degree of disability
[13]. Over the years, it has also been found that when EEG paroxysmal abnormalities recur less frequently than typical ESES, cognitive disturbances can appear, but in a more heterogeneous manner and tending to be less severe than typical ESES. According to several authors, the current trend is to consider a spike and wave index (SWI) > 60% as suggestive of CSWS
[14]. Even according to the current classification of the International League Against Epilepsy, the 85% threshold of SWI is no longer mandatory for the diagnosis of CSWS
[5]. However, the defining features and diagnostic criteria of CSWS, and in particular of ESES, are still a matter of debate. Further, for a long time, the terms CSWS and ESES have been used interchangeably, but currently, ESES mainly refers to the EEG pattern, while CSWS refers to the syndromic picture associated with ESES; even on this point, however, there is no unanimity of opinion
[15]. The main purpose of therapy for CSWS should be to prevent or reduce cognitive deficits
[16]. Data regarding epidemiology are scarce, but CSWS cases are assumed to be 0.5% to 0.6% of children with epilepsy followed at tertiary referral epilepsy centers
[17]. It is probable, however, that this recurrence is underestimated
[18]. Etiopathogenesis, when known, is heterogeneous. To give an idea, Sonnek et al.
[19], in a large retrospective monocentric study (95 children), also including “near CSWS” patients (SWI = 40–85%), found a structural/metabolic etiology in 43.2% of cases and genetic alterations in 17.9%, while the etiology was unknown in 38.9%. Regarding the pathogenetic mechanisms, it has been hypothesized that the negative effects of continuous EEG paroxysmal abnormalities on cognitive functions and behavior may be related to the spike-induced damage of the synaptic homeostasis processes that occur physiologically in sleep and that are crucial during development. During sleep, homeostasis is favored by synaptic weakening/elimination following the synaptic strength increase during wakefulness. Alterations in synaptic strength are shown in EEGs through alterations of sleep slow-wave activity (SWA). During CSWS, there are no sleep SWA changes, which occur again after CSWS remission: this finding suggests a spike-related impairment of the synaptic homeostasis during sleep. All of this may impair cortical wiring, causing the neuropsychological dysfunctions that are typical of CSWS
[20][21][20,21].
2. Continuous Spike–Waves during Slow Sleep
2.1. EEGs and Clinics of Typical and Atypical ESES
The core features of typical ESES, as described by Tassinari et al.
[12], are (1) electrical-status epilepticus lasting for ≥85% of NREM (slow) sleep, documented by more than two EEG recordings during a period of ≥1 month; (2) different degrees of global cognitive deterioration, with decrease in full-scale IQ and performance IQ
[26]; (3) behavioral disorders, including attention deficit, hyperactivity, aggressiveness, difficulty in social interaction, and (more rarely) psychosis (autistic behavior is also possible)
[12][27][28][29][12,27,28,29]; (4) epileptic seizures, focal or apparently generalized, with a heterogeneous semeiology including absences; epileptic falls; clonic, tonic–clonic, partial motor, and complex partial seizures; and negative myoclonus (see atonic component for example at the upper limbs, showed through polygraphic recordings or restricted to lower limbs) can be associated in atypical absences
[30][31][32][30,31,32]; it is possible that there are no overt clinical seizures
[33], but this is an infrequent occurrence
[19][34][35][19,34,35]; and (5) motor signs, including dyspraxia, ataxia, and dystonia. The described clinical EEG characteristics have substantially remained the same over the years
[21]. Overnight EEG is considered as the gold standard for ESES diagnosis
[36] because an ESES pattern may be recorded during the first sleep cycle of the night and may appear fragmented in subsequent ones. But in clinical practice, even an ambulatory EEG can be effective, according to several authors. For example, Nagyova et al. retrospectively studied 199 consecutive pediatric patients who underwent an ambulatory EEG. Suspected ESES was the motivation in 38.6% of cases, and this examination was useful in 97.5% of cases
[37].
In the atypical forms of ESES, there is a great heterogeneity in the reported clinical features depending on the frequency and localization of the EEG paroxysmal abnormalities
[38]. In fact, the localization of prevailing paroxysmal abnormalities is more heterogeneous than in the typical forms (focal, multifocal, unilateral, asymmetric, or symmetric bilateral), and diffuse paroxysmal abnormalities have been reported
[39].
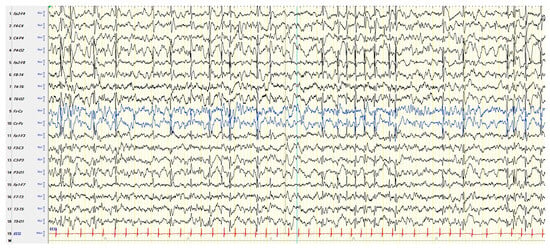
Figure 1 shows an example of atypical ESES.
Figure 1. NREM sleep EEG in a boy aged 5 years and 7 months with autism spectrum disorder, intellectual disability, and focal epilepsy, showing subcontinuous diffuse spike–waves (lasting < 85% of NREM sleep), prevailing in the right posterior regions. NREM: nonrapid eye movement. EEG: electroencephalogram.
Among the neuropsychiatric clinical features in atypical ESES, ADHD-like symptoms are prominent and seem to be related positively to SWI
[40]. Acquired visual agnosia may be found in CSWS, as suggested in the case reported by Van Iterson et al.: a male aged almost 7 years presenting CSWS, prevailing in the left posterior regions, who lost the skill of recognizing familiar faces and naming pictures
[41]. Kuki et al. reported acquired Kanji (morphogram) dysgraphia associated with visual processing impairment in two children with CSWS prevailing in the occipito-temporal region, in which functional neuroimaging showed a dysfunction located in the left posterior temporal lobe
[42]. According to Tassinari et al. (also in individual cases), an adequate neurophysiological study aiming at localizing the “functional lesion”, associated with a detailed neuropsychological assessment, could help to analyze the cortical networks responsible for specific cognitive functions
[43]. However, not all authors agree on the existence of a clear correlation between EEG picture and neuropsychological impairment, such as Pavlidis et al., who studied 24 children with idiopathic encephalopathy related to ESES (three had LKS). They found a lack of correlation between the severity/type of cognitive impairment and SWI and/or a topography of EEG paroxysmal abnormalities, suggesting the possible role of further factors during sleep (including an altered sleep homeostasis) in the neurobehavioral disorders
[44]. It should not be forgotten that any structural brain abnormalities, if they are not the primary cause of the CSWS picture, can still influence its characteristics. For example, Mohammadi et al. described one child with hemi-ESES (that is, limited to one cerebral hemisphere) related to a congenital corpus callosum absence, inhibiting CSWS propagation from the right hemisphere to the left one. Their findings point out the focal origin of ESES as well as the corpus callosum’s role in the bilateral synchronous expression of EEG paroxysmal abnormalities
[45]. On the other hand, the 85% SWI limit that separates typical forms of ESES from atypical ones may be of relative importance
[5]. Caraballo et al., in a multicenter, retrospective long-term follow-up study including 117 CSWS cases, found similar electroclinical features comparing patients with a >85% SWI and patients with a <85% SWI
[46]. Gencpinar et al. retrospectively analyzed the EEG recordings of 44 ESES/CSWS cases with a follow-up of at least two years in order to study (a) the SWI during NREM sleep EEG (>85% in 33 cases (typical ESES) and =50–85% in 11 (atypical ESES)) and (b) the maximum amplitude area of CSWS: anterior in 33 cases and posterior in 11. The authors found that the SWI rate (typical versus atypical ESES) as well as the maximum amplitude area of CSWS (anterior versus posterior) were not significantly related to clinical and brain imaging features or to the response to treatment (see seizure control and SWI reduction)
[47]. Particularly in atypical forms of ESES, the technique called magnetoencephalography, utilizing superconducting quantum interference devices, can be useful in precisely localizing the spike source
[48]. The diagnosis of CSWS, on the electrophysiological level, is therefore based on qualitative and quantitative criteria whose detection in the past was entrusted solely to the human eye. Over the years, increasingly precise and reliable automatic systems have been developed for the detection and quantification of spike–waves. These systems are very useful in CSWS, where EEG paroxysmal abnormalities are particularly frequent and their classic visual quantification can be very tiring
[49]. Yu et al. proposed an SWI quantification neural network (SQNN) using a pre-labeling algorithm in order to quantify SWI automatically in children with ESES. The authors found that it would be possible to accurately and reliably quantify SWI through the SQNN, with a processing speed 100 times faster than that of experts
[50]. However, to quantify the number of EEG paroxysmal abnormalities, SWI computing is not the only method: in fact, the number of spikes per unit of time can be also calculated. It should be borne in mind that the “time occupied by epileptiform activities” (i.e., SWI) and the “number of spikes per unit of time”, otherwise known as the “spike wave frequency” (SWF), differ substantially. Using the SWF, researchers can avoid the ceiling effect that is intrinsic to SWI calculation, especially when EEG paroxysmal abnormalities are more than 60 per minute, because SWI cannot exceed the limit of 100%, while SWF has no defined upper limit. Unfortunately, it is unclear whether the SWF or the SWI is more related to ESES clinical features
[51][52][51,52]. In any case, the SWI is still the most used method today to quantify the number of EEG paroxysmal abnormalities. Not using the same method does not help with data sharing among CSWS researchers. Apart from this last important aspect, in general, the lack of studies sharing the same terminology concerning CSWS can be an obstacle to the reciprocal communication among clinicians and researchers dealing with this condition
[53]. Still regarding the diagnosis on the electrophysiological level, Carvalho et al. showed that a wearable EEG device with only two bipolar channels for 24 h, through a semiautomatic template-match spike search, could be a good alternative, less expensive and better tolerated, to the full 10–20 long-term ambulatory EEG that is the classic tool for interictal spike quantification. In a clinical context, this alternative could be useful, particularly for the spike activity follow-up
[54].
CSWS syndrome can begin as such, but very often it is the electroclinical evolution of another, more or less severe, epilepsy. For example, it can follow Ohtahara syndrome, a severe early-onset form of epileptic encephalopathy
[55], but the most frequent occurrence is CSWS as the evolution of Rolandic epilepsy, otherwise known once as benign epilepsy with centro-temporal spikes
[38] and today, with a more correct expression, as self-limited epilepsy with centro-temporal spikes (SeLECTS) (see also later). This is very important to keep in mind, as it shows us the possibility of changing from one apparently benign form of epilepsy to another with a less favorable prognosis. Further, CSWS may be the atypical electroclinical evolution of Panayiotopoulos syndrome, as suggested in two cases reported by Oguni et al.
[56]. The recurrence of CSWS has also been described in a rare epileptic condition, sunflower epilepsy, a photosensitive and usually drug-resistant reflex epilepsy with seizures characterized by one’s head turning towards light, similarly to a sunflower, and a hand waving in front of the eyes
[57]. The neuropsychiatric outcome is often disappointing
[51][58][59][51,58,59]. Margari et al. carried out a long-term follow-up study in 25 CSWS children. At the onset of CSWS, 96% of the cases showed one or more neuropsychiatric disorders, including behavioral problems in 54%, intellectual disability in 37.5%, learning disorders in 33%, developmental coordination disorder in 17%, language disorder in 12.5%, and autism spectrum disorder in 8%. In the course of the follow-up, neuropsychiatric disorders persisted unaltered in 52% of the cases, worsened in 24%, and improved in 24%. Most of the cases without improvement in the follow-up had symptomatic CSWS
[60].
How can CSWS interfere with cognitive functions? Researchers know that spikes shown in EEGs correspond topographically to focal cortical areas of hypermetabolism according to the results of the positron emission tomography (PET)
[61]. In healthy individuals, the physiologic overnight decrease in the slow-wave slope in NREM sleep is closely related to the sleep recovery function that is necessary for optimal cognitive performance. Starting from this assumption, Bölsterli Heinzle et al. retrospectively analyzed 14 CSWS patients, identifying the spike–wave “focus” as the area of the highest spike amplitude and the frequency of spike–waves as the SWI. They found no overnight change in the slow-wave slope in the “focus” are, while in “nonfocal” areas, the slow-wave slope decreased significantly. The spike–wave density (see SWI) was correlated with the overnight slope decrease impairment: the higher the SWI, the more impaired the slope decrease. The authors concluded by proposing that the overnight slope decrease impairment is a possible mechanism leading to CSWS neuropsychological deficits
[62]. In another study, Bölsterli et al. carried out a retrospective analysis of overnight EEG in 10 children with idiopathic ESES. Throughout the night, the slope of the slow waves did not diminish significantly during ESES, especially at the focus of paroxysmal abnormalities, while after ESES remission, the slope diminished significantly. Compared to healthy controls, cases after ESES remission showed no significant difference in overnight slope decrease. The best cognitive outcome after ESES remission has been found in three cases who had some degree of slope decrease during ESES. These findings suggest that alterations in NREM-sleep slow waves induced by ESES are reversible, and the severity of cognitive impairment might be related to the severity of slow-wave impairment during ESES. Therefore, slow-wave analysis might be a prognostic factor for cognitive outcomes
[63]. The longitudinal course of the slow-wave slope’s overnight change could be an objective EEG marker related to the cognitive function course
[64]. Through a retrospective cross-sectional study considering 22 CSWS cases, Bölsterli Heinzle et al. found that the maximal spike–wave location, corresponding to the epileptic focus, was age-related and followed a posterior–anterior trajectory, similarly to various focal epilepsies. Therefore, younger cases should be more likely to show posterior foci than older ones. The authors hypothesized that this posterior–anterior trajectory of CSWS maximal spike–waves could be related to maturational modifications to the maximal expression of sleep slow waves
[65]. Moreover, EEG paroxysmal abnormalities during NREM sleep, causing both an activation in epileptogenic zones (particularly in perisylvian and in prefrontal areas) and a deactivation of the default mode network (DMN) located beyond the epileptogenic area, may produce neuropsychological impairments
[66]. For perisylvian network epilepsies, including idiopathic focal childhood epilepsies, ESES, and LKS, Halász and Szűcs proposed that a sleep-related alteration in physiological neural networks may underlie epileptogenesis. Homeostatic plasticity, which is a compensative process for cell loss recovery, may be associated with an excitability increase up to an epileptic level. In this way, physiological functioning derails to a pathological (epileptic) working mode. NREM sleep heightens epileptic processes, which in turn impair sleep functions. The vicious cycle between sleep and epilepsy works every night, altering brain functions, particularly during development. The type and degree of cognitive impairment should be related to the involved network’s function
[67]. Ng and Hodges found that, in a comparison with patients with other types of epilepsy, children with recently diagnosed ESES turned out to be less impaired on the neurocognitive level than children with generalized epilepsy. This finding suggests that neuropsychological differences between ESES and other epileptic syndromes may develop as a long-term consequence of the neurological disorder and/or of pharmacological treatment
[68]. However, within generalized epilepsies, there can be very heterogeneous situations in terms of severity and clinical characteristics, so much so as to make it difficult to interpret the data obtained by these authors
[69].
Table 1 summarizes the clinical and EEG features of typical and atypical ESES.
Table 1.
Clinical and EEG features of typical and atypical ESES.
| |
Clinics |
EEG |
Typical
ESES |
Cognitive deterioration.
Behavioral disorders, including attention deficit, hyperactivity, aggressiveness, difficulty in social interaction, and (more rarely) psychosis;
autistic behavior is also possible.
Epileptic seizures, focal or apparently generalized, with a heterogeneous semeiology.
Motor signs, including dyspraxia, ataxia, and dystonia. |
Electrical-status epilepticus lasting
for ≥85% of NREM (slow) sleep,
documented by more than 2 EEG
recordings during a period of ≥1
month. |
Atypical
ESES |
Great heterogeneity in the reported clinical features depending on the frequency and localization of the EEG paroxysmal abnormalities: a cognitive deterioration has been reported, but less frequently than typical ESES.
Behavioral disorders, in particular ADHD-like symptoms.
Epileptic seizures, focal or apparently generalized, with a heterogeneous semeiology. |
Paroxysmal abnormalities lasting
for <85% of NREM sleep
(usually > 50% < 85%), with a localization
more heterogeneous than typical
forms: focal, multifocal, unilateral,
asymmetric or symmetric bilateral,
and diffuse. |
2.2. Predictive Factors of the Evolution into CSWS
Considering the relevant impact of CSWS on cognitive neurodevelopment, it is very important to recognize, if possible, what the predictive factors of an evolution into CSWS are. Rolandic epilepsy is the prototype of benign epilepsies in childhood, but in some cases, it may have an atypical development, including CSWS. A retrospective study by Pesántez-Ríos et al. comprised nine patients with SeLECTS, all of whom showed atypical clinical features and CSWS. The average age at onset of Rolandic seizures was 5 years, while clinical and EEG deterioration occurred, on average, one and a half years later. Electroclinical features suggesting an atypical-development SeLECTS were the early onset of seizures; the appearance of new seizures with increased frequency; and the presence of an EEG fronto-centrotemporal focus, with increasing frequency, both in wakefulness and in sleep
[70]. Porat Rein et al. carried out a retrospective cohort study involving 104 cases with SeLECTS. They identified the following risk factors for the development in these patients of severe atypical variants, including LKS and ESES: EEG spike–waves; EEG without evidence of left lateralization; and centro-temporal, frontal, or fronto-temporal EEG localization
[71]. Subsequently, Porat Rein et al. developed a predictive model of the transformation of SeLECTS into epileptic encephalopathy with CSWS or LKS, collecting data from a cohort of 91 SeLECTS cases, of which 18 showed an encephalopathic transformation. The most important risk factors were the localization of EEG paroxysmal abnormalities in fronto-temporal and temporo-parietal areas and seizure semiology, including dysarthria or somatosensory auras
[72]. But other studies were not limited to investigating the possible evolution into CSWS of SeLECTS alone. Desprairies et al. carried out a retrospective case–control study to search for any clinical or EEG feature indicative of later CSWS development at the time of the first seizure. They included 10 CSWS cases with available EEG at the time of the first seizure compared with 10 matched controls. They found no clinical or EEG features suggesting later CSWS development. However, during the follow-up, the occurrence of multiple types of seizures and seizure worsening were significantly more frequent in the CSWS cases
[73]. According to Caraballo et al., clinical parameters predicting an evolution into CSWS were early-onset seizures, the appearance of new seizures, and a relevant increase in seizure frequency, while EEG parameters predicting an evolution into CSWS may be an increased frequency of the interictal EEG paroxysmal abnormalities during wakefulness and sleep, and the presence of bilateral spikes and waves
[74]. Aeby et al., through a retrospective study, compared the awake EEG of 15 CSWS patients (including one with LKS: see later) with 15 matched cases with self-limited focal epilepsy (SFE). They found that, at the time of cognitive regression, CSWS patients were more likely than SFE cases to have a slow-wave index (SLWI) > 6%, an SWI > 10%, a cluster of spike–waves (CLSW) of ≥1 s, an SWF of >11%, and an EEG score of ≥3 (qualitative assessment: from grade 0 (normal) to grade 4 (maximally pathological))
[75].
Table 2 summarizes the clinical and EEG predictive factors of the evolution into CSWS.
Table 2.
Predictive factors of the evolution into CSWS.
| Clinical |
Early onset of seizures; multiple types of seizures; appearance of new seizures with an increased frequency; seizure semiology including dysarthria or somatosensory auras. |
| EEG |
Fronto-centro-temporal focus, with increasing frequency, both in wakefulness and in sleep; pattern EEG of spike–waves. |