1. Introduction
Apoptosis is a highly regulated form of programmed cell death that plays a crucial role in various physiological and pathological conditions, including development, tissue homeostasis, and the removal of damaged or unnecessary cells
[1][2][3][4][1,2,3,4]. It is now well established that defects in the apoptotic pathways are closely related to both oncogenesis and cancer treatments resistance
[5][6][7][8][5,6,7,8]. Understanding the molecular mechanisms regulating apoptosis is therefore of crucial importance for the identification of specific targets for anticancer therapies. Apoptosis execution relies on the highly regulated activation of a group of cysteine proteases called caspases that specifically cleave a series of substrates, resulting in cell death
[9][10][11][9,10,11]. Caspases are synthesized as inert zymogens that are activated through two distinct, but interconnected, pathways, called the intrinsic or extrinsic pathways, in which apoptotic stimuli trigger the activation of the so-called initiator caspases (such as caspase-2, -8, -9, and -10) which, in turn, proteolytically cleave and activate effector (or executioner) caspases (caspase-3, -6, and -7)
[12][13][12,13]. When activated, the effector caspases specifically cleave a broad spectrum of cellular targets, ultimately leading to cell death.
The extrinsic pathway is initiated by the activation of death receptors, upon binding of their cognate ligands and subsequent recruitment at the level of the cytoplasmic region of the death receptors of death domain-containing adaptor proteins
[2]. This results in the formation of a death-inducing signaling complex (DISC), which can in turn recruit and activate caspase-8 via oligomerization. Death receptor-mediated apoptosis can be inhibited by a proteolytically inactive homolog of caspase-8, called cellular FLICE inhibitory protein (cFLIP), which can be recruited to the DISC, forming a proteolytically inactive heterodimer with caspase-8
[14].
The intrinsic pathway, also known as the mitochondrial pathway, proceeds through the induction of the mitochondrial outer membrane permeabilization (MOMP) and the subsequent release in the cytoplasm of numerous proapoptotic mitochondrial constituents
[15]. Among these, cytochrome
c promotes the oligomerization of apoptotic protease-activating factor-1 (APAF-1), triggering the formation of the apoptosome and dimerization-induced activation of caspase-9
[16]. The intrinsic pathway is intricately regulated by pro- and anti-apoptotic B-cell lymphoma-2 (Bcl-2) family members, which consist of evolutionarily conserved proteins that share at least one Bcl-2 homology (BH) domain
[17].
Although the connection between the number of genetic mutations and cancer is complex, the tumorigenesis process relies on both the activation of oncogenes that stimulate cancer cells proliferation and survival, as well as the inactivation of tumor suppressor genes that hold cellular proliferation in check
[18]. To date, a wide variety of oncogenes and tumor suppressor genes involved in the regulation of pro- or anti-apoptotic signals have been discovered. Variations in the expression of these genes, or their mutation, can contribute to tumor initiation, progression or resistance to treatment. Consequently, a number of therapeutic approaches have been developed to overcome cell death resistance through the pharmacological manipulation of various apoptosis signaling networks. Current main therapeutic strategies include either inhibiting antiapoptotic regulators or stimulating proapoptotic factors
[19][20][19,20]. For instance, a number of inhibitors of antiapoptotic Bcl-2 family members, which are known to be overexpressed in numerous cancers, are now used in clinics. These include the Bcl2-selective BH3-mimetic Venetoclax
[21], which is currently used for the treatment of chronic lymphocytic leukemia, small lymphocytic lymphoma, or acute myeloid leukemia, or the myeloid cell leukemia-1 (Mcl-1) inhibitors S63845, AMG-176, and AZD5991
[22][23][24][22,23,24].
Among the cell death regulators is API5 (apoptosis inhibitor-5), also known as AAC-11 (anti-apoptosis clone 11 or FIF (fibroblast growth factor-2 interacting factor)), a 55 kDa nuclear scaffold protein initially discovered as a negative regulator of apoptosis upon nutritional stress conditions
[25]. API5 has emerged as a key player in the context of cancer as its overexpression has been associated with aggressive tumor behavior, resistance to treatment, and poor prognosis
[26][27][28][29][30][31][32][33][34][35][26,27,28,29,30,31,32,33,34,35]. Furthermore, recent observations indicate that API5 influence extends far beyond apoptosis regulation, making this intriguing protein a versatile regulator of cell fate with diverse functions ranging from anti-apoptosis to metastasis, cell cycle control, mRNA export, and TLR4-dependent activation and maturation of antigen presenting cells. API5’s intricate involvement in these critical cellular processes underscores its significance in both health and disease, particularly in cancer biology.
2. Apoptosis Inhibitor 5 and Cancer
2.1. API5 Expression and Prognosis Value
An expression analysis of API5 revealed an ubiquitous but varying expression of API5 in cancers. Of note, the human
API5 gene is located in chromosomal segment 11p12-13, in a region that is amplified in a number of cancers
[36][37][38][39][111,112,113,114]. API5 has been shown to be upregulated in various cancers, such as breast cancer, colorectal cancer, cervical cancer, NSCLC, or B-cell chronic lymphoid leukemia
[26][28][29][30][31][32][33][35][40][41][42][26,28,29,30,31,32,33,35,86,115,116]. This expression appears to be clinically relevant as it is associated with poor overall and disease-free survival as well as resistance to treatment, suggesting a potential role for API5 as a prognosis and survival marker
[26][28][29][30][31][32][33][35][40][26,28,29,30,31,32,33,35,86]. In line with this hypothesis, Cho and colleagues have shown that API5 expression levels gradually increased during the normal-to-tumor transition of cervical carcinoma
[28]. As API5 overexpression has been linked to cancer cell proliferation (see above) and survival (see below), one might envision that API5 could contribute to the development and progression of cancer.
2.2. API5’s Role on Cancer Metastasis, Immune Response, and Survival
The widespread and high expression of API5 in tumors suggests that API5 contributes to human malignancy. Interestingly, a recent study has shown that overexpression of API5 in breast epithelial cells induces a partial epithelial–mesenchymal transition (EMT)-like phenotype
[40][86]. EMT is a differentiation process through which transformed epithelial cells gain the ability to invade and disseminate
[43][117]. In line with this hypothesis, API5 expression has been demonstrated to contribute to tumor invasion and metastases in various cancer settings
[26][29][34][40][26,29,34,86]. One important step in invasion is the remodeling and disassembly of the extracellular matrix and its constituents through enzymes such as matrix metalloproteinases (MMPs)
[44][118]. MMPs are structurally related, zinc-dependent endopeptidases that have been linked to a wide variety of pathological states, including carcinogenesis, and elevated levels of MMPs correlate with unfavorable prognosis in multiple cancers
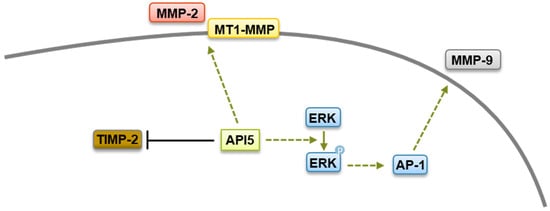
[45][119]. An API5-forced expression increases levels of MMP-2 as well as membrane type 1 matrix metalloproteinase (MT1-MMP), with concomitant downregulation of the tissue inhibitor of MMP (TIMP-2)
[29]. While the mechanism by which API5 regulates MMP-2 and MT1-MMP expression are not clear yet, API5 expression has been linked to the upregulation of the transcriptional coactivator β-catenin, which is well known to possess a crucial role in cell invasion and to regulate MMPs expression
[46][120]. Furthermore, using other tumor settings, Song and colleagues demonstrated that API5 enhanced MMP-9 expression through an ERK-dependent regulation of activator protein 1 (AP-1)
[34]. Therefore, it is possible that API5 contributes to cancer metastasis through β-catenin- and ERK-mediated MMPs expression (
Figure 1).
Figure 1. Metastasis regulation by API5. API5 increases tumor cell metastasis via upregulation of MMP-2, MMP-9, and MT1-MMP expression and downregulation of TIMP-2 levels.
Another mechanism by which API5 contributes to tumor progression is through the induction of tumor immune escape. Indeed, in a very interesting study, the group led by Tae Woo Kim demonstrated that API5 plays key roles in both tumor progression and immunity
[31]. Using different murine cancer models, Kim and colleagues showed that API5 could render tumor cells resistant to immune-mediated cytotoxicity, through the inhibition of tumor-specific T-cell-mediated apoptosis
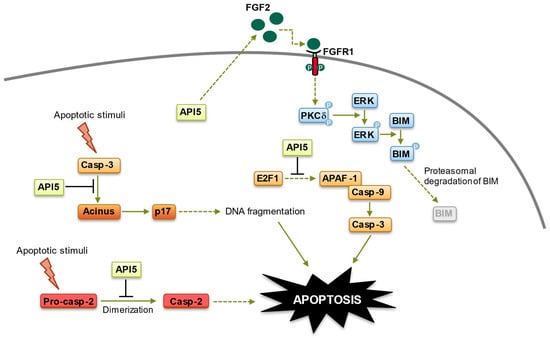
[31]. Mechanistically, API5 hinders T-cell-triggered apoptosis of cancerous cells by the upregulation of FGF-2 and subsequent activation of the FGFR1–PKCδ–ERK pathway, resulting in the ubiquitin-dependent degradation of the BH3-only protein BIM (
Figure 2). Although these observations need to be confirmed using primary samples, they fit well with previous data indicating that cancer cells with high levels of AKT/ERK exhibit suppressed BIM expression
[47][48][121,122], and they identify API5 as an immune-related prognostic biomarker. Therapeutic targeting of API5 could therefore represent a potential treatment option for cancer through tumor immune escape.
Figure 2. Anti-apoptotic functions of API5. API5 regulation of apoptosis takes place at four levels: (1) API5 inhibits E2F1-induced apoptosis. (2) API5 inhibits Acinus-induced apoptotic DNA fragmentation. (3) API5 inhibits caspase-2 activation. (4) API5 upregulates FGF2/FGFR1 signaling, leading to BIM degradation. Of note, API5-mediated activation of FGFR1 signaling, which triggers proteasome-dependent degradation of BIM, is also involved in the chemo- and immune-resistance of cancer cells.
Finally, a substantial number of studies have shown that API5, which was initially identified for its antiapoptotic function, is critically involved in tumor survival and resistance to chemotherapeutic drugs. Morris and colleagues initially noted that the depletion of API5 was specifically lethal to tumor cells with deregulated E2F1
[49][39]. Shortly after, a crucial role for API5 in tumor cells’ sensitivity to anticancer drug was demonstrated by different groups. Indeed, the silencing of API5 in various cell lines sharply increased tumor cells’ sensitivity to chemotherapeutic drugs such as etoposide, camptothecin, or cisplatin, whereas API5-forced expression endowed cancer cells with enhanced resistance to these agents
[26][50][51][26,65,123]. Although more research is needed to completely decipher the mechanisms at play, API5-induced resistance to anticancer drugs has been shown to stem from its activation of the FGFR1 signaling, which triggers the ERK-mediated degradation of the proapoptotic protein BIM, as well as the inhibition of caspase-2 and apoptotic DNA fragmentation
[26][50][51][52][26,51,65,123] (
Figure 2). Recently, API5 silencing has been linked to a sharp increase in cell death of caspase 9
−/− Jurkat cells treated with ABT-263, a potent and selective inhibitor of Bcl-2 and Bcl-xL
[53][124]. Interestingly, ABT-263 is known to synergize with chemotherapies inducing DNA damage
[54][55][125,126]. As the silencing of API5 promotes ABT-263-induced DNA damage
[53][124], it is possible that API5 could function as a regulator of the DNA repair machinery, as its association with the chromatin remodeler ALC1 (amplified in liver cancer 1), which plays a key role in DNA repair, suggests
[56][49]. In line with this hypothesis, API5 has been shown to be upregulated by UV irradiation of primary liver cells, and an increased expression of API5 protects primary liver cells from UV-induced apoptosis and to increase glioblastoma cells to radioresistance
[57][58][59][127,128,129].
Combined, these data demonstrate a crucial role for API5 in cancer cell development and progression, providing a rationale for the therapeutic targeting of API5 for cancer treatment.
2.3. Targeting API5 as a Therapeutic Approach
Cancer is a consequence of multiple deregulated processes that endow tumor cells with certain traits which were described as “Hallmarks of Cancer” by Hanahan and Weinberg two decades ago
[60][130]. Numerous new potential cancer targets have been identified over the last few years, and survival pathways, angiogenesis, DNA damage response (DDR), senescence pathways or the immune system, for instance, are important types of targets for the development of anticancer drugs. Clearly, given the above-described functions of API5, targeting this intriguing protein could be of great interest for cancer treatment. Among the different opportunities to indirectly or directly target proteins are their inhibition at the expression level, their inhibition through physical degradation or their inhibition at the protein/protein interaction level.
The downregulation of API5 expression has been achieved so far by means of RNA interference (RNAi), short hairpin RNAs (shRNAs), or microRNA (miRNA), and all these approaches have demonstrated interesting potentialities as they have resulted in cancer cells death, increased sensitivity to anticancer agents or immune-mediated cytotoxicity or inhibition of metastasis potential (see above). Therefore, RNA-based therapeutics approaches for API5 expression targeting could open novel possibilities for cancer treatment. However, critical challenges in applying these RNA therapies, related to pharmacodynamics and pharmacokinetics as well as immunogenicity issues, have hindered the clinical progress of RNA-based drugs
[61][131]. Nonetheless, a substantial number of RNA-based therapeutics are currently under clinical investigation for various diseases, including cancers, and several RNA-based medications have been approved for clinical use
[62][132]. Therefore, further research on RNA-based therapeutics for API5 targeting might lead to more RNA-based therapeutics for cancer treatment.
Direct API5 degradation is another therapeutic option. API5 stability has been demonstrated to be regulated via acetylation at lysine 251 (K251) by the histone acetyltransferase p300, which leads to an increase inAPI5 stability, whereas deacetylation by the histone deacetylase 1 HDAC1 reduces API5 levels
[63][53]. Consequently, chemical inhibition of p300 resulted in decreased API5 levels, affecting its functions in cell cycle
[63][53]. Interestingly, the expression of an acetylation-deficient mutant of API5 (K251A) did not protect tumor cells from apoptosis induced by serum deprivation
[64][40]. Furthermore, tumor cells expressing API5 K251A in an API5 knockdown background could not survive while in culture
[63][53]. Therefore, one can envision that the use of p300 inhibitors could constitute an interesting therapeutic option for the induction of the direct degradation of API5. As a matter of fact, the development of p300 inhibitors has attracted great attention in recent years due to its potential therapeutic value in the treatment of cancers
[65][66][133,134]. Consequently, the steady-state API5 acetylation–methylation equilibrium, which functions as a molecular rheostat governing API5 stability and antiapoptotic properties, might be amenable to therapeutic exploitation as an anti-cancer strategy.
Finaly, the inhibition of API5 interactions with its partner proteins is another approach to target its biological functions. API5 interacts with several apoptosis-related proteins, and this complex-forming ability—probably favored by its elongated 3D structure
[64][40]—appears to be essential for API5 to fulfill its antiapoptotic or metastatic functions
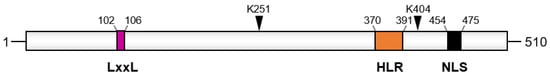
[25][34][50][56][67][25,34,48,49,65]. Among its different domains, API5 contains several protein–protein interaction modules, such as HEAT and ARM repeat or the heptad leucine repeat region (
Figure 3). Researchers has shown that the heptad leucine repeat region of API5 mediates its interaction with several of its partners, such as Acinus and the kinase p21-activated kinase 1 (PAK1)
[50][68][54,65]. Moreover, mutations of conserved residues (leucines 384 and 391 or arginine 382) in this domain abrogate API5 biological functions and prevent its interaction with its molecular partners
[25][34][50][66][67][68][25,34,48,54,65,134]. This indicates that the heptad leucine repeat region of API5 could constitute a therapeutic target for anti-cancer drugs. Researchers have constructed two API5-derived cell-permeable peptides, called RT53 and RT39, that comprise portions of the heptad leucine repeat region of API5 fused to a cell-penetrating sequence
[50][68][54,65]. Both peptides acted as decoys and were able to prevent interaction between API5 and Acinus or PAK1
[50][68][54,65]. Moreover, the peptides demonstrated potent pro-apoptotic activity and synergy with anticancer drugs, as well as anti-migration potential, on multiple cancer cell lines as well as primary cutaneous T-cell lymphoma (Sézary syndrome) cells, thus phenocopying the consequences of API5 silencing
[50][68][69][70][71][54,65,135,136,137]. The peptides also demonstrated in vivo efficacy as single agents in murine models of melanoma, breast cancer, acute promyelocytic leukemia, and Sézary syndrome, with very favorable half-lives in mice
[27][50][68][69][70][71][72][73][27,54,65,135,136,137,138,139]. Structurally, RT53 and RT39 adopt a helical conformation, with an N-terminal stretch of arginine and lysine residues followed by a hydrophobic region, making them amphipathic and membrane active, similarly to other know membranolytic peptides
[74][140]. Therefore, while sparing normal cells, the RT53 and RT39 peptides also possess oncolytic properties
[68][69][70][71][73][54,135,136,137,139]. Mechanistically, RT39 retention in the membrane of Sézary cells is dependent on binding to PAK1 at the level of the plasma membrane, where PAK1 is strongly expressed
[68][69][54,135]. Interestingly, oncolysis mediated by RT53 exhibited the hallmarks of immunogenic cell death, and vaccines consisting of APL or melanoma cells exposed in vitro to RT53 induced prophylactic and therapeutic protection in syngeneic murine models
[70][71][136,137]. Therefore, RT53 and RT39 peptides’ anti-cancer action stems from both their ability to prevent API5 biological functions, through protein–protein interaction inhibition, and through their oncolytic properties.
Figure 3. Domain organization of API5. The LxxLL motif, the acetylation site (K251), the SUMOylation site (K404), the heptad leucine repeat (HLR), and the nuclear localization domain (NLS) are shown. Numbers indicate amino acids positions.
Recently, the crystal structure of the API5–FGF-2 complex has been solved, allowing for the determination of the precise domains of the proteins involved in their interaction
[75][47]. Based on this knowledge, Bong and colleagues have developed lentiviruses expressing a peptide composed of API5 residues 183–191, a domain involved in API5 interaction with FGF-2. Interestingly, the lentivirus-mediated expression of the API5-derived peptide in HeLa cells abrogated API5–FGF-2 interaction and reduced the nuclear export of bulk RNA, which is dependent on the API5–FGF-2 complex
[75][47]. While it remains to be determined whether this novel API5-derived peptide exhibits anticancer effect or synergizes with anticancer drugs, these data support the testing of API5-derived peptides for cancer treatment.