Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Joyeeta Roy.
The estrogen receptor (ER) plays a critical role in the initiation and progression of breast cancer. Utilizing specialized therapies aimed at the ER has been effective in many instances and is commonly employed in breast cancer treatment protocols. The selection of therapy depends on multiple factors, including the menopausal status, breast cancer stage, and unique tumor attributes. These therapies can function independently as monotherapy, or in conjunction or sequential alignment with other treatments, based on the distinct characteristics of the breast cancer and the patient’s overall health.
- breast cancer
- estrogen receptor
- endocrine therapy
1. Introduction
Breast cancer continues to be the most prevalent type of cancer among women, constituting to roughly 25% of all newly diagnosed cases and 16% of global cancer-related deaths. Regrettably, advanced breast cancer that has metastasized to distant organs is beyond a cure with the treatments currently available. According to the American Cancer Society, projections for the US in 2023 indicate that approximately 297,790 women will be diagnosed with invasive breast cancer and about 43,700 will succumb to the disease. Advancements in the treatment of breast cancer have decreased death rates by 43% from 1989 to 2020, translating to 460,000 less breast cancer deaths during that time [1]. The surrogate intrinsic subtypes are typically used clinically and are based on histology and immunohistochemistry expression; the subtypes of breast cancer have been identified as Luminal A-like, Luminal B-like HER2−, Luminal B-like HER2+, HER2-enriched, and Triple-negative. Luminal A-like accounts for 60–70% of breast cancer cases in the United States, followed by Luminal B-like HER2− at 10–20%, triple negative at 10–15%, and HER2-enriched and Luminal B-like HER2+ together at approximately 13–15% (Figure 1) [2,3][2][3]. Among the different types, Luminal A tumors, the most prevalent molecular subtype, tend to grow at a slower pace compared to the other cancer types. Luminal A breast cancers are a subtype of breast cancer that are typically hormone-receptor-positive (ER/PR-positive), HER2-negative, low proliferation and a low-risk GES (gene expression signature). This means they generally respond well to endocrine therapy (ET) and have a better prognosis compared to other subtypes [4]. By contrast, the Luminal B-like group is HR-positive but have an ER and PR expression lower than Luminal A-like, but it exhibits high grade or high proliferation and high-risk GESs. Whereas TNBC and non-luminal types demonstrate aggressive characteristics such as high grade, lack of ER, PR and HER2 expression, and high proliferation, making them a distinct breast cancer subtype tied to poorer prognoses [5]. Despite numerous attempts to find potential therapeutic targets, chemotherapy continues to be the primary systemic treatment for TNBC. The development of specialized agents for TNBC has not progressed as much as those targeting ER and HER2 in other clinical subtypes. However, substantial recent progress has been made with innovative treatments, such as poly adenosine diphosphate (ADP)-ribose polymerase inhibitors (PARPi) for patients with germline BRCA1/2 mutations. These inhibitors were approved by the FDA in 2019 for metastatic disease and later in 2021 for early stages of the disease [6,7,8][6][7][8]. The first immunotherapy regimens for TNBC were also approved for metastatic disease beginning in 2019 [9] and for early disease in 2021 [10]. The HER2-enriched (HR−/HER2+) subtype was associated with the worst prognosis in the past. However, the use of targeted therapies for HER2+ cancers has significantly improved outcomes for these patients. Trastuzumab, which was introduced 25 years ago, has become the standard of care treatment and has greatly improved the treatment of HER2+ breast cancer patients [11]. Trastuzumab functions by attaching to the IV domain of the HER2 receptor, which blocks downstream signaling and triggers both antibody-dependent cell-mediated cytotoxicity and antibody-dependent cell-phagocytosis. Presently, Trastuzumab is given alongside chemotherapy or radiotherapy for one year in an adjuvant setting. In a neo-adjuvant context, Trastuzumab is combined with Pertuzumab, a monoclonal antibody that aims at the II domain of the HER2 molecule, suppressing its ability to dimerize [12]. However, despite Trastuzumab showing improved responses and outcomes, a significant portion of patients still develop resistance to the therapy and experience a recurrence of the disease [13]. Given these clinical results, considerable endeavors have been made to develop new therapies targeting HER2. These include monoclonal antibodies targeting different HER2 epitopes, antibody-drug conjugates, bispecific antibodies, tyrosine kinase inhibitors (TKIs), and others [14]. Efforts are also ongoing to stimulate the immune response in HER2+ patients, following the evident benefits derived from immunotherapy in TNBC. Numerous strategies are being utilized to achieve this goal, such as the administration of checkpoint inhibitors, the connection of effector T cells with HER2 antibodies, the implementation of cellular therapy, and the use of vaccines [3,15][3][15]. As for HR+ breast cancer treatment, endocrine therapy remains a cornerstone, but improvements in patient outcomes have been achieved by introducing novel therapeutic agents that act on extra-hormonal molecular targets—such as cyclin-dependent kinase 4/6 (CDK4/6), the mammalian target of rapamycin (mTOR), protein kinase B (also known as Akt), and phosphatidylinositol 3-kinase (PI3K). However, advanced HR+ breast cancer still deems uncurable. It often manifests through mechanisms that are independent of hormonal signaling, leading to recurrence, treatment resistance, and potential metastasis, thus accentuating the necessity for additional translational research.

Figure 1. Surrogate intrinsic subtypes, standard of care and prognosis of breast cancer.
2. Endocrine Therapy: Treatment for ER+/HER2− MBC
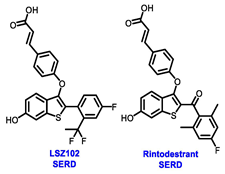
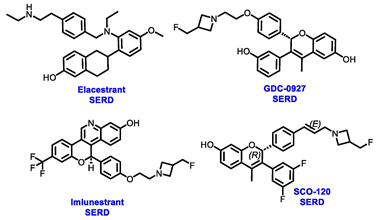
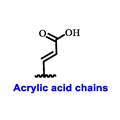
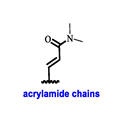
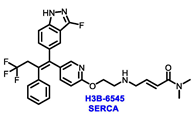
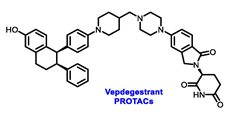
Endocrine therapy is typically categorized into three classes. The first class of drugs used are SERMs, with tamoxifen serving as a prominent example. SERMs act as competitive inhibitors of estrogen-ER binding and display either agonist or antagonist activity, depending on the target tissue. These drugs function by inhibiting the binding of estrogens and preventing ER signaling. While SERMs are effective at blocking the activation function AF2 domain, they are incapable of blocking the AF1 domain of ERα, which can lead to agonist activity and limit its effectiveness [17,18][16][17]. The second class comprises the 3rd generation aromatase inhibitors (AIs), which function by inhibiting estradiol biosynthesis, thereby preventing ER signaling. Nonsteroidal AIs, such as anastrozole and letrozole, function as noncovalent, reversible competitive inhibitors of androgen–aromatase binding. Conversely, the steroidal AI, such as exemestane, covalently and irreversibly binds to the aromatase substrate-binding site [19][18]. The third class is the SERDs, of which fulvestrant and elacestrant are the current approved drugs in this category [20,21][19][20]. SERDs are high-affinity competitive antagonists that block ER dimerization and DNA binding, inhibit nuclear uptake, and increase the turnover and degradation of the ER. This overall leads to an improved inhibition of estrogen signaling. CDK4/6 inhibitors are often used in conjunction with AIs or SERDs to enhance efficacy, particularly for advanced or metastatic breast cancer (MBC). To enhance efficacy, especially for advanced or metastatic breast cancer, CDK4/6 inhibitors are often used in tandem with AIs or SERDs. It has been evidenced that combining CDK4/6 inhibitors with endocrine therapy boosts the objective response rate (ORR), progression-free survival (PFS), and overall survival (OS) in ER positive MBC. This improvement was observed when CDK4/6 inhibitors were added to an AI for first-line ET or fulvestrant as a second-line ET, after progression or relapse on an AI [22,23,24][21][22][23]. The approval of fulvestrant and elacestrant represent significant advancements as pure ERα antagonist drugs and led the discovery of a new class of anti-estrogen drugs, including CERANs, SERCAs, and PROTACs that target ER (Figure 2, Table 1). CERANs work by impeding all agonist signaling facilitated by ERα through the total inactivation of the ER by suppressing both activation functions of ER transcription (AF-1 and AF-2). As a result, they antagonize the ER, block transcriptional activity, and promote ER degradation [25][24]. On the other hand, SERCAs contain a Michael acceptor (e.g., arylamide chain) to attach covalently to a cysteine residue (C530) of the ER, thereby functioning as covalent inhibitors of ER transcription (Figure 2) [26][25]. PROTACs function by binding both an E3 ubiquitin ligase and a target protein concurrently; thus, bringing the ubiquitin ligase complex into proximity to the target protein. This proximity enables the transfer of ubiquitin from E2 to the target protein, thus triggering its eventual degradation by the proteasome (Figure 2). In recent years these innovative approaches have emerged as a leading modality in drug discovery [27,28,29][26][27][28].
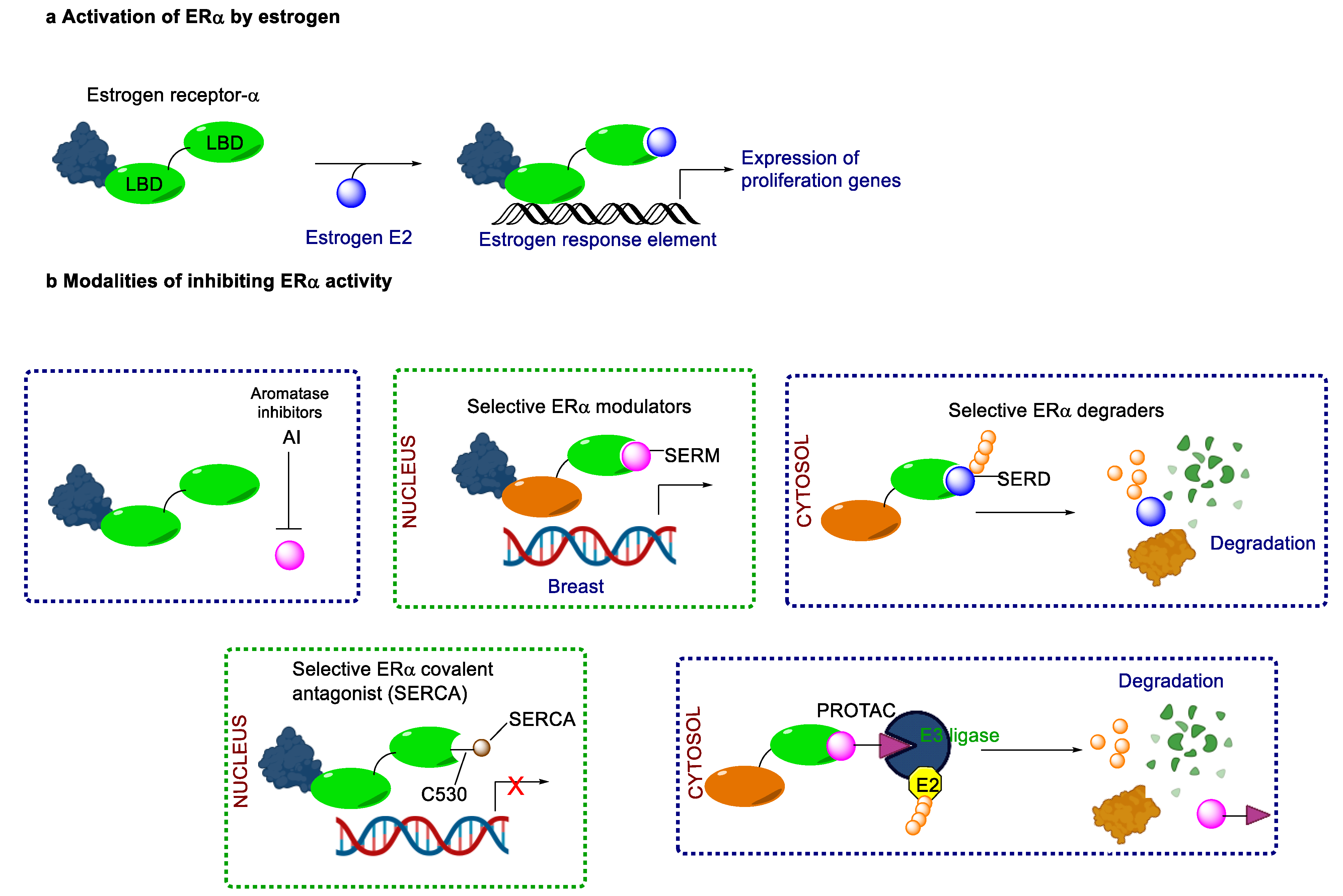
Figure 2. Estrogen receptor-α signaling and modes of inhibition.
Table 1. Structures of new generation Endocrine agents (SERDs, SERCA, CERAN and PROTACs).
| ERα Ligand Core | Side Chain | Structures of SERDs, SERCA, CERAN and PROTACs |
|---|---|---|
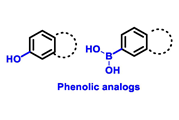 |
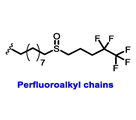 |
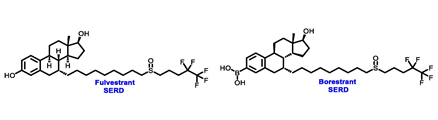 |
[34]. To improve the prediction of resistance, there is a need for more extensive, real-time monitoring of the dynamic mutational patterns, both during and after treatment. This necessitates employing methods that involve examining additional prognostic biomarkers like ESR1, PIK3CA, and AKT mutations. Real-time monitoring can be implemented through diverse techniques, such as liquid biopsy methods that analyze circulating tumor DNA (ctDNA) or other liquid biopsy markers. These strategies offer a non-invasive option and potentially enable more frequent evaluations of the tumor’s genetic makeup, allowing for timely adjustments to the treatment plan in response to evolving genomic alterations.
Table 2. Prevalence of ESR1 mutations in ctDNA and outcomes in metastatic breast cancer.
3. Prevalence of ESR1 Mutations in Metastatic Breast Cancer
Endocrine therapy is typically successful in treating the majority of patients with hormone receptor-positive (HR+) advanced breast cancer. However, over time, cancer cells may develop resistance to this therapy, either by acquiring new mutations or losing hormone receptor expression [30][29]. Primary endocrine resistance is typically characterized by a relapse within the first two years of adjuvant endocrine therapy or disease progression within the initial six months of endocrine therapy for advanced or metastatic breast cancer. On the other hand, secondary or acquired resistance is identified as a relapse that transpires after a minimum of two years of endocrine therapy, either during or within the first year of completing adjuvant endocrine therapy [4,31][4][30].
ESR1 mutations represent one of the most well-known and extensively investigated molecular mechanisms of therapeutic resistance. These mutations can cause ligand-independent activity, fostering tumor growth and resistance to ET. The prevalence of such mutations depends on the duration of ET and can be found in 20%–40% of patients previously treated with aromatase inhibitors (AIs) for MBC. However, these mutation rates are much lower in the case of recurrent breast cancer and less than 1% in ET-naive patients. This suggests that ESR1 mutations may be acquired during AI treatment in the metastatic setting [32][31]. In the PADA-1 trial, it was observed that patients with pre-existing ESR1 mutations undergoing AI and CDK4/6 inhibitor therapy for MBC experienced an increase in the ESR1 mutation, reaching up to 27% at a median time of 15.6 months (Table 2) [33][32]. In the BOLERO-2 clinical trial, which enrolled 541 patients, 29% of participants (156 patients) exhibited a mutation in the estrogen receptor. Interestingly, patients with the mutation had a significantly shorter median OS (20.7 months) than those without the mutation (32.1 months) [34][33]. Other established ET resistance mechanisms include the growth-promoting PI3K-AKT-mTOR and RAS/RAF/MEK/ERK pathways [35]
4. Data from Early Clinical Trials and Lessons Learned
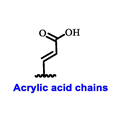
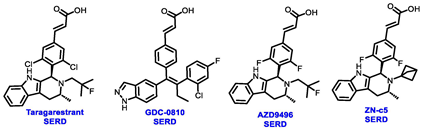
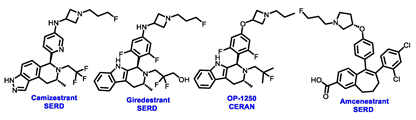
Multiple early-phase clinical trials have explored oral SERDs featuring acrylic acid side chains, including GDC-0810, AZD9496 and LSZ102 (Table 1) [38][37]. These agents have demonstrated significant antitumor activity in both endocrine-sensitive and resistant preclinical models, as well as in ESR1-mutated tumors [39,40,41,42,43][38][39][40][41][42]. Despite their potential, early-phase clinical trials have unveiled challenges indicating lower efficacy and tolerability, leading to the limited progression of most of these compounds beyond phase I.
For example, the clinical trial (NCT02569801) of GDC-0810 was terminated due to its inferior effectiveness compared to fulvestrant in a phase II study. Despite its ability to degrade ERα significantly, reaching 91%, GDC-0810 lacks a complete antagonist profile and weakly activates ER transcription, potentially impacting in vivo efficacy [44][43]. The clinical trials for AZD9496 have shown low response rates and an unfavorable toxicity profile. Considering the modest clinical benefits and unfavorable toxicity profile, the development of AZD9496 was halted in February 2021, opting instead for its more potent and well-tolerated successor, camizestrant [45][44]. Likewise, the phase I clinical trial of LSZ102 (NCT02734615) was discontinued due to interim single-agent results, indicating an ORR of only 1.3% in heavily pretreated ER+ metastatic breast cancer patients. However, when LSZ102 was used in combination with ribociclib, a CDK4/6 inhibitor, alpelisib, or a PI3K inhibitor, the overall response rate improved to 17% or 7%, respectively [46,47][45][46].
Several additional SERDs demonstrating preclinical activity are under evaluation in phase I–II clinical trials, including taragarestrant (NCT03471663) [48][47], ZN-c5 (NCT03560531) [49][48], GDC-0927 (NCT02316509) [50][49], SCO-120 (NCT04942054) [48][47]. But unfortunately, the degradative properties did not translate into a therapeutic benefit, leading to its discontinuation or halt in development; this highlights the challenges in creating new molecular entities that encompass the full set of desirable features.
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48.
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874.
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66.
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513.
- Liao, M.; Zhang, J.; Wang, G.; Wang, L.; Liu, J.; Ouyang, L.; Liu, B. Small-Molecule Drug Discovery in Triple Negative Breast Cancer: Current Situation and Future Directions. J. Med. Chem. 2021, 64, 2382–2418.
- Tutt, A.N.J.; Garber, J.E.; Kaufman, B.; Viale, G.; Fumagalli, D.; Rastogi, P.; Gelber, R.D.; de Azambuja, E.; Fielding, A.; Balmana, J.; et al. Adjuvant Olaparib for Patients with BRCA1- or BRCA2-Mutated Breast Cancer. N. Engl. J. Med. 2021, 384, 2394–2405.
- Robson, M.E.; Tung, N.; Conte, P.; Im, S.A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann. Oncol. 2019, 30, 558–566.
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763.
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59.
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821.
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20.
- Hurvitz, S.A.; Martin, M.; Symmans, W.F.; Jung, K.H.; Huang, C.S.; Thompson, A.M.; Harbeck, N.; Valero, V.; Stroyakovskiy, D.; Wildiers, H.; et al. Neoadjuvant trastuzumab, pertuzumab, and chemotherapy versus trastuzumab emtansine plus pertuzumab in patients with HER2-positive breast cancer (KRISTINE): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2018, 19, 115–126.
- Perez, E.A.; Romond, E.H.; Suman, V.J.; Jeong, J.H.; Davidson, N.E.; Geyer, C.E., Jr.; Martino, S.; Mamounas, E.P.; Kaufman, P.A.; Wolmark, N. Four-year follow-up of trastuzumab plus adjuvant chemotherapy for operable human epidermal growth factor receptor 2-positive breast cancer: Joint analysis of data from NCCTG N9831 and NSABP B-31. J. Clin. Oncol. 2011, 29, 3366–3373.
- Mercogliano, M.F.; Bruni, S.; Mauro, F.L.; Schillaci, R. Emerging Targeted Therapies for HER2-Positive Breast Cancer. Cancers 2023, 15, 1987.
- Swain, S.M.; Shastry, M.; Hamilton, E. Targeting HER2-positive breast cancer: Advances and future directions. Nat. Rev. Drug Discov. 2023, 22, 101–126.
- Howell, A.; Cuzick, J.; Baum, M.; Buzdar, A.; Dowsett, M.; Forbes, J.F.; Hoctin-Boes, G.; Houghton, J.; Locker, G.Y.; Tobias, J.S.; et al. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for breast cancer. Lancet 2005, 365, 60–62.
- Francis, P.A.; Pagani, O.; Fleming, G.F.; Walley, B.A.; Colleoni, M.; Lang, I.; Gomez, H.L.; Tondini, C.; Ciruelos, E.; Burstein, H.J.; et al. Tailoring Adjuvant Endocrine Therapy for Premenopausal Breast Cancer. N. Engl. J. Med. 2018, 379, 122–137.
- Miller, W.R.; Bartlett, J.; Brodie, A.M.H.; Brueggemeier, R.W.; Di Salle, E.; Lonning, P.E.; Llombart, A.; Maass, N.; Maudelonde, T.; Sasano, H.; et al. Aromatase inhibitors: Are there differences between steroidal and nonsteroidal aromatase inhibitors and do they matter? Oncologist 2008, 13, 829–837.
- Bross, P.F.; Cohen, M.H.; Williams, G.A.; Pazdur, R. FDA drug approval summaries: Fulvestrant. Oncologist 2002, 7, 477–480.
- Bhatia, N.; Thareja, S. Elacestrant: A new FDA-approved SERD for the treatment of breast cancer. Med. Oncol. 2023, 40, 180.
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936.
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2- Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884.
- Slamon, D.J.; Neven, P.; Chia, S.; Jerusalem, G.; De Laurentiis, M.; Im, S.; Petrakova, K.; Valeria Bianchi, G.; Martin, M.; Nusch, A.; et al. Ribociclib plus fulvestrant for postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer in the phase III randomized MONALEESA-3 trial: Updated overall survival. Ann. Oncol. 2021, 32, 1015–1024.
- Hodges-Gallagher, L.; Sun, R.; Myles, D.; Klein, P.; Zujewski, J.A.; Harmon, C.; Kushner, P. OP-1250: A potent orally available complete antagonist of estrogen receptor-mediated signaling that shrinks wild type and mutant breast tumors. Eur. J. Cancer 2020, 138, S55.
- Rioux, N.; Smith, S.; Korpal, M.; O’Shea, M.; Prajapati, S.; Zheng, G.Z.; Warmuth, M.; Smith, P.G. Nonclinical pharmacokinetics and in vitro metabolism of H3B-6545, a novel selective ER covalent antagonist (SERCA). Cancer Chemother. Pharm. 2019, 83, 151–160.
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114.
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. Drug Development. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381.
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559.
- Huppert, L.A.; Gumusay, O.; Idossa, D.; Rugo, H.S. Systemic therapy for hormone receptor-positive/human epidermal growth factor receptor 2-negative early stage and metastatic breast cancer. CA Cancer J. Clin. 2023, 73, 480–515.
- Cardoso, F.; Paluch-Shimon, S.; Senkus, E.; Curigliano, G.; Aapro, M.S.; Andre, F.; Barrios, C.H.; Bergh, J.; Bhattacharyya, G.S.; Biganzoli, L.; et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann. Oncol. 2020, 31, 1623–1649.
- Will, M.; Liang, J.; Metcalfe, C.; Chandarlapaty, S. Therapeutic resistance to anti-oestrogen therapy in breast cancer. Nat. Rev. Cancer 2023, 23, 673–685.
- Bidard, F.C.; Hardy-Bessard, A.C.; Dalenc, F.; Bachelot, T.; Pierga, J.Y.; de la Motte Rouge, T.; Sabatier, R.; Dubot, C.; Frenel, J.S.; Ferrero, J.M.; et al. Switch to fulvestrant and palbociclib versus no switch in advanced breast cancer with rising ESR1 mutation during aromatase inhibitor and palbociclib therapy (PADA-1): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2022, 23, 1367–1377.
- Chandarlapaty, S.; Chen, D.; He, W.; Sung, P.; Samoila, A.; You, D.; Bhatt, T.; Patel, P.; Voi, M.; Gnant, M.; et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncol. 2016, 2, 1310–1315.
- Juric, D.; Janku, F.; Rodon, J.; Burris, H.A.; Mayer, I.A.; Schuler, M.; Seggewiss-Bernhardt, R.; Gil-Martin, M.; Middleton, M.R.; Baselga, J.; et al. Alpelisib Plus Fulvestrant in PIK3CA-Altered and PIK3CA-Wild-Type Estrogen Receptor-Positive Advanced Breast Cancer: A Phase 1b Clinical Trial. JAMA Oncol. 2019, 5, e184475.
- Spoerke, J.M.; Gendreau, S.; Walter, K.; Qiu, J.; Wilson, T.R.; Savage, H.; Aimi, J.; Derynck, M.K.; Chen, M.; Chan, I.T.; et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat. Commun. 2016, 7, 11579.
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. J. Clin. Oncol. 2016, 34, 2961–2968.
- Wu, Y.L.; Yang, X.; Ren, Z.; McDonnell, D.P.; Norris, J.D.; Willson, T.M.; Greene, G.L. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol. Cell 2005, 18, 413–424.
- Joseph, J.D.; Darimont, B.; Zhou, W.; Arrazate, A.; Young, A.; Ingalla, E.; Walter, K.; Blake, R.A.; Nonomiya, J.; Guan, Z.Y.; et al. The selective estrogen receptor downregulator GDC-0810 is efficacious in diverse models of ER plus breast cancer. Elife 2016, 5, e15828.
- Lai, A.; Kahraman, M.; Govek, S.; Nagasawa, J.; Bonnefous, C.; Julien, J.; Douglas, K.; Sensintaffar, J.; Lu, N.; Lee, K.J.; et al. Identification of GDC-0810 (ARN-810), an Orally Bioavailable Selective Estrogen Receptor Degrader (SERD) that Demonstrates Robust Activity in Tamoxifen-Resistant Breast Cancer Xenografts. J. Med. Chem. 2015, 58, 4888–4904.
- De Savi, C.; Bradbury, R.H.; Rabow, A.A.; Norman, R.A.; de Almeida, C.; Andrews, D.M.; Ballard, P.; Buttar, D.; Callis, R.J.; Currie, G.S.; et al. Optimization of a Novel Binding Motif to (E)-3-(3,5-Difluoro-4-((1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-2,3,4,9-tetrahydro-1H-pyridoindol-1-yl)phenyl)acrylic Acid (AZD9496), a Potent and Orally Bioavailable Selective Estrogen Receptor Downregulator and Antagonist. J. Med. Chem. 2015, 58, 8128–8240.
- Andreano, K.J.; Wardell, S.E.; Baker, J.G.; Desautels, T.K.; Baldi, R.; Chao, C.A.; Heetderks, K.A.; Bae, Y.; Xiong, R.; Tonetti, D.A.; et al. G1T48, an oral selective estrogen receptor degrader, and the CDK4/6 inhibitor lerociclib inhibit tumor growth in animal models of endocrine-resistant breast cancer. Breast Cancer Res. Treat. 2020, 180, 635–646.
- Tria, G.S.; Abrams, T.; Baird, J.; Burks, H.E.; Firestone, B.; Gaither, L.A.; Hamann, L.G.; He, G.; Kirby, C.A.; Kim, S.; et al. Discovery of LSZ102, a Potent, Orally Bioavailable Selective Estrogen Receptor Degrader (SERD) for the Treatment of Estrogen Receptor Positive Breast Cancer. J. Med. Chem. 2018, 61, 2837–2864.
- Guan, J.; Zhou, W.; Hefner, M.; Blake, R.A.; Chalouni, C.; Chen, I.P.; De Bruyn, T.; Giltnane, J.M.; Hartman, S.J.; Heidersbach, A.; et al. Therapeutic Ligands Antagonize Estrogen Receptor Function by Impairing Its Mobility. Cell 2019, 178, 949–963.
- Paoletti, C.; Schiavon, G.; Dolce, E.M.; Darga, E.P.; Carr, T.H.; Geradts, J.; Hoch, M.; Klinowska, T.; Lindemann, J.; Marshall, G.; et al. Circulating Biomarkers and Resistance to Endocrine Therapy in Metastatic Breast Cancers: Correlative Results from AZD9496 Oral SERD Phase I Trial. Clin. Cancer Res. 2018, 24, 5860–5872.
- Jhaveri, K.; Juric, D.; Yap, Y.S.; Cresta, S.; Layman, R.M.; Duhoux, F.P.; Terret, C.; Takahashi, S.; Huober, J.; Kundamal, N.; et al. A Phase I Study of LSZ102, an Oral Selective Estrogen Receptor Degrader, with or without Ribociclib or Alpelisib, in Patients with Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2021, 27, 5760–5770.
- Ferraro, E.; Walsh, E.M.; Tao, J.J.; Chandarlapaty, S.; Jhaveri, K. Accelerating drug development in breast cancer: New frontiers for ER inhibition. Cancer Treat. Rev. 2022, 109, 102432.
- Rej, R.K.; Thomas, J.E., 2nd; Acharyya, R.K.; Rae, J.M.; Wang, S. Targeting the Estrogen Receptor for the Treatment of Breast Cancer: Recent Advances and Challenges. J. Med. Chem. 2023, 66, 8339–8381.
- Keogh, G.P.; Papish, S.; Piskorski, W.; Ulanska, M.; Jackson, B.; Suster, M.; Ptaszynski, M.; Mina, L. A phase Ib dose-escalation study of ZN-c5, an oral selective estrogen receptor degrader (SERD), in combination with abemaciclib in patients with advanced estrogen receptor (ER)+/HER2-breast cancer. Ann. Oncol. 2021, 32, S618–S619.
- Dickler, M.N.; Villanueva, R.; Fidalgo, J.A.P.; Mayer, I.A.; Boni, V.; Winer, E.P.; Hamilton, E.P.; Bellet, M.; Urruticoechea, A.; Gonzalez-Martin, A.; et al. A first-in-human phase I study to evaluate the oral selective estrogen receptor degrader (SERD), GDC-0927, in postmenopausal women with estrogen receptor positive (ER+) HER2-negative metastatic breast cancer (BC). Cancer Res. 2018, 78, PD5-10.
More