Glioblastoma (GBM) is the most aggressive and common form of primary brain cancer with a dismal prognosis. Current GBM treatments have not improved patient survival, due to the propensity for tumor cell adaptation and immune evasion, leading to a persistent progression of the disease. In recent years, the tumor microenvironment (TME) has been identified as a critical regulator of these pro-tumorigenic changes, providing a complex array of biomolecular and biophysical signals that facilitate evasion strategies by modulating tumor cells, stromal cells, and immune populations. Efforts to unravel these complex TME interactions are necessary to improve GBM therapy. Immunotherapy is a promising treatment strategy that utilizes a patient’s own immune system for tumor eradication and has exhibited exciting results in many cancer types; however, the highly immunosuppressive interactions between the immune cell populations and the GBM TME continue to present challenges.
- glioblastoma
- immunotherapy
- tumor microenvironment
- biophysics
1. Introduction
2. Biophysical Aspects of the TME
2.1. ECM Composition
Solid tumors comprise a wide range of highly expressed ECM proteins that include laminins, fibronectin, elastin, and fibrillar collagen [63][48]. These proteins provide functionality and stability to the tumor environment for disease progression and accounts for up to 60% of the tumor mass [64][49]. Indeed, the presence of these proteins plays a key role in regulating the pro-invasion and therapeutic resistance within the tumor that help to direct cell migration, adhesion, and proliferation similar to that seen in early development [65][50]. Several ECM proteins of interest in GBM have been identified over the last several years including osteopontin, hyaluronic acid (HA), and laminin and are known to increase invasion potential through mechanotransduction signals transmitted throughout the cytoskeleton [66,67,68][51][52][53]. Further, HA is the primary ECM component in the brain, and it has been strongly implicated in GBM tumor development and depends on the HA molecular weight—high-molecular-weight HA has an anti-tumor effect and low-molecular-weight HA has a pro-tumor effect [67,69,70,71,72][52][54][55][56][57].2.2. ECM Mechanics
Besides ECM composition, the mechanics of the ECM can direct cell behavior [73][58] and tumor malignancy. Increased substrate stiffness has been shown to increase proliferation and single-cell migration in GBM. ECM stiffness can also increase cell adhesiveness, which interestingly reduces collective migration due to dense networks of cell–substrate adhesions [74,75][59][60]. These stiffnesses also affect the density of the matrix and can facilitate primary tumor escape during metastasis [76][61] by providing permissive porous environments to increase cell escape from the tumor or inhibitory dense environments that would confine the cell to the tumor.2.3. Interstitial Fluid Flow
The increased mechanics of the intratumoral space caused by the high stiffness and density can also affect the flow of interstitial fluid throughout the tumor. The flow of interstitial fluid through the tumor exposes the cells to fluid pressure and soluble cues such as pro-angiogenic factors and anti-inflammatory TGF-β1 signals [79][62]. These factors function to inhibit anti-tumor immune cells and promote pro-tumor immune cells by maintaining the inflammation of the tumor to a sustainable level.3. Biophysical TME-Immune Cell Interactions
It has been established that the biophysics of the TME and ECM can regulate cell behavior in a pro-tumorigenic capacity. However, besides cancer cells, other types of cells including the immune cell populations are regulated by the biophysical TME (Figure 1).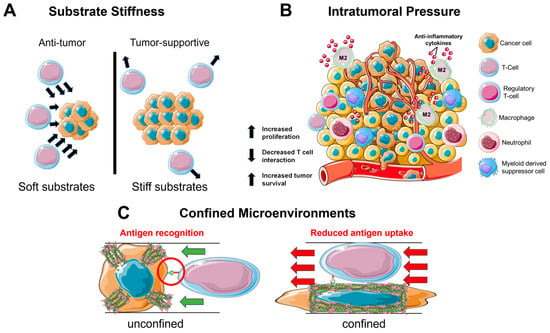
3.1. Substrate Stiffness
3.2. Intratumoral Pressure
In a similar manner to the stiff tumor, an interstitial fluid flow induces pro-tumorigenic effects on the immune cells in the TME (Figure 1B). Tumors often utilize the lymphatic system to invade and metastasize [99][68]. This system specializes in fluid drainage and, therefore, it is not uncommon to find heightened pressure gradients at the junction of tumor and lymphatics. These junctions experience an increased flow and mechanical stress, which serves to cause DNA damage and increase the production of cytokines such as TGF-β1 to reduce inflammation [99,100,101][68][69][70]. Furthermore, the interstitial pressure is sufficient to induce a pro-tumorigenic macrophage phenotype as seen by the upregulation of M2 macrophage markers ArgI, TGM2, and CD206 [102][71]. This development of M2 macrophages further works to inhibit T cell function via the additional TGF-β1 production and expression of immune checkpoint ligands that inactivate infiltrating T cells [103,104][72][73].3.3. Confined Migration
Considering the density of the brain parenchyma, it is unsurprising that GBM is an innately aggressive cancer. For many cell types, migration in a confined environment can lead to DNA damage via nuclear rupture [104][73], which serves as the basis for the development of metastatic cancer [111][74]. During infiltration of the TME, the immune cells will enter a state of confined migration (Figure 1C). Macrophages have been observed to form a protective actin cortex that shields against compressive forces that can damage the nucleus and lead to cell death [112][75]. This enables a more efficient and safe form of migration necessary for patrolling throughout the confined environments. Likewise, T cells navigate a variety of confined spaces as they patrol throughout the body; however, while patrolling, the T cells also interact with antigen-presenting cells. This presents a balance that must be maintained between migrating quickly through an environment and spending enough time in the same location as the antigen-presenting cell to become activated against an antigen [113][76].4. Bioengineering Systems
4.1. Biomaterial Systems
Biomaterials are synthetic or natural materials that can be engineered to mimic physiological and pathological environments and are foundational to investigate cell-ECM interactions. Among the most common biomaterials are hydrogel-culture-based systems due to their versatility in controlling factors such as stiffness, ECM composition, and other biomolecules such as growth factors [122][77]. In the context of solid tumors such as GBM, the most important immune cell functions to consider are tumor infiltration and tumor cell elimination. Hydrogel-based platforms have proven vital in dissecting the interplay between immune cells and the tumor ECM. The mechanosensing capacity of T cells has been interrogated via TCR-mediated activation through the introduction of HA binding [123][78] and the tuning of the stiffness to biomimetic levels [123,124][78][79].4.2. Microfabrication
Microfabrication approaches such as 3D printing and microfluidics represent important tools for studying the contribution of geometric cues as well as observing cell phenomena in a single-cell context. Many studies leverage these tools to decipher the migration mechanisms of immune cells. In the 3D context, CD8+ T cell amoeboid migration operates through a contractility driven mechanism via RhoA activation that is unique to the 2D environment [129,130,131][80][81][82]. Additionally, the T cell transfection efficiency can be modified on microfluidic systems through mechanoporation via stretching to engineer T cells with greater motility, antigen recognition, and antigen elimination [132,133][83][84].4.3. Advanced In Vitro Systems
Creating more complex, biologically relevant in vitro systems serves as an intermediate step before reaching in vivo models. Co-culture models combine multiple cell types into a single culture environment to analyze real-time interactions between the cells, and lab-on-a-chip technology is useful for combining multiple physiological systems to determine how cell processes differ between the environmental changes. Macrophages and microglia are normally susceptible to reprogramming via GBM cues that promote tumor development; yet, when cultured separately, these changes cannot be observed. Therefore, co-culture systems allow for the necessary paracrine and juxtacrine interactions (ephs/ephrins [137][85] or P-selectin/PSGL-1 [138][86]) involved in the bidirectional communication between the tumor and immune cells to occur and be observed. These interactions are intended to help recognize the cancer cells for immune-cell-mediated elimination but are often dysregulated to instead promote tumor survival and development.5. Immunotherapy Applications
From the mechanistic insights gained through bioengineering systems, researchers can leverage these interactions to optimize immunotherapy protocols to increase efficacy and improve patient survival. By engineering the culture expansion platforms of immune cells and the activities of immune cells, efforts to increase the tumoricidal capacity of immune cells are growing. In recent years, many types of cancers have seen significant improvement due to the development of novel immunotherapies, their developments in treating GBM, and the biophysical design of the therapies (Figure 2).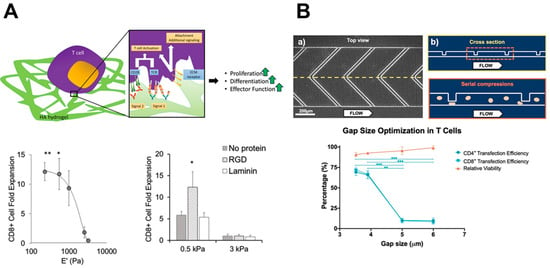
5.1. Tunable Hydrogel Culture Systems
5.2. Chimeric Antigen Receptor Therapy
In addition to the removal and reintroduction of cells following the ex vivo differentiation into an anti-tumor phenotype, the direct modification of immune cells has also had success in treating various types of cancers. The generation of chimeric antigen receptors (CARs) for T cells involves removing T cells from a patient and using gene engineering to induce the production of a single type of synthetic receptor for antigen recognition [149,150][90][91]. To increase successful T cell transfection, microfluidic systems have stretched cells to increase membrane pore formation, which allows for greater mRNA entry to generate the CAR (Figure 2B) [132,133][83][84].5.3. Checkpoint Blockade
Another therapy option utilized in even more cancers is the use of immunologic checkpoint blockades. This therapy uses antibodies to inhibit the programmed death of T cells induced by the binding of PD-1 or to prevent the activation of the T cell via CTLA-4 [160,161,162][92][93][94]. These receptors naturally become activated to prevent T cell overactivity; however, cancer cells can trigger these pathways as well to limit T cell-mediated tumor killing through the reduction in cytokine and granule production. To prevent this inhibition, antibodies are developed that are specific to either the tumor cell (PD-L1) or the T cell (PD-1/CTLA-4) and are administered [160,161,162,163][92][93][94][95]. The type of antibody used and its receptor target are variable depending on the state of the tumor and the effectiveness in promoting T cell activity.6. Conclusions
GBM is an extremely deadly type of brain cancer that has remained difficult to treat in part due to its high resistance to standard therapies and immunotherapy. The dynamic TME is a key driver of cancer progression and presents pro-malignant biophysical signals including ECM composition, density, stiffness, and interstitial fluid flow, which helps direct the flow of soluble cues, increases the intertumoral pressure on the cells, and generates hypoxic regions. Ultimately, these factors work together to promote tumor growth and disease progression; however, these factors also dysregulate immune cells through the production of immunosuppressive cytokines, the recruitment of inhibitory immune cells, and/or the conversion into tumor-supportive tumor-associated immune cells. Biomedical research has made strides in elucidating the underlying mechanisms of the biophysical TME on immunosuppression through novel bioengineered tools and platforms. The insights gained have been eagerly applied to bolster immune cell expansion techniques and enhance immunotherapy through the biophysical manipulation of the ECM and immune cell receptor manipulation.References
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507.
- Ohgaki, H.; Kleihues, P. Population-Based Studies on Incidence, Survival Rates, and Genetic Alterations in Astrocytic and Oligodendroglial Gliomas. J. Neuropathol. Exp. Neurol. 2005, 64, 479–489.
- Ostrom, Q.T.; Price, M.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2015–2019. Neuro-Oncology 2022, 24 (Suppl. 5), v1–v95.
- Stupp, R.; Hegi, M.E.; Mason, W.P.; Van Den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466.
- Delgado-López, P.D.; Corrales-García, E.M. Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071.
- Omuro, A.; DeAngelis, L.M. Glioblastoma and Other Malignant Gliomas: A Clinical Review. JAMA 2013, 310, 1842–1850.
- Li, J.; Feng, L.; Lu, Y. Glioblastoma multiforme: Diagnosis, treatment, and invasion. J. Biomed. Res. 2023, 37, 47.
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma—Are we there yet? Neuro-Oncology 2013, 15, 4–27.
- Birzu, C.; French, P.; Caccese, M.; Cerretti, G.; Idbaih, A.; Zagonel, V.; Lombardi, G. Recurrent glioblastoma: From molecular landscape to new treatment perspectives. Cancers 2020, 13, 47.
- Jiang, H.; Yu, K.; Li, M.; Cui, Y.; Ren, X.; Yang, C.; Zhao, X.; Lin, S. Classification of progression patterns in glioblastoma: Analysis of predictive factors and clinical implications. Front. Oncol. 2020, 10, 590648.
- Rapp, M.; Baernreuther, J.; Turowski, B.; Steiger, H.-J.; Sabel, M.; Kamp, M.A. Recurrence pattern analysis of primary glioblastoma. World Neurosurg. 2017, 103, 733–740.
- Brandes, A.A.; Tosoni, A.; Franceschi, E.; Sotti, G.; Frezza, G.; Amistà, P.; Morandi, L.; Spagnolli, F.; Ermani, M. Recurrence pattern after temozolomide concomitant with and adjuvant to radiotherapy in newly diagnosed patients with glioblastoma: Correlation with MGMT promoter methylation status. J. Clin. Oncol. 2009, 27, 1275–1279.
- Ståhl, P.; Henoch, I.; Smits, A.; Rydenhag, B.; Ozanne, A. Quality of life in patients with glioblastoma and their relatives. Acta Neurol. Scand. 2022, 146, 82–91.
- Hodi, F.S.; O’day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723.
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813.
- Ribas, A. Releasing the brakes on cancer immunotherapy. N. Engl. J. Med. 2015, 373, 1490–1492.
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 2018, 379, 2108–2121.
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F. Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. N. Engl. J. Med. 2018, 378, 2078–2092.
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S. Neoadjuvant PD-1 blockade in resectable lung cancer. N. Engl. J. Med. 2018, 379, 2185.
- Hanna, N.; Burton, R.C. Definitive evidence that natural killer (NK) cells inhibit experimental tumor metastases in vivo. J. Immunol. 1981, 127, 1754–1758.
- Chan, I.S.; Ewald, A.J. The changing role of natural killer cells in cancer metastasis. J. Clin. Investig. 2022, 132.
- Teng, M.W.; Galon, J.; Fridman, W.-H.; Smyth, M.J. From mice to humans: Developments in cancer immunoediting. J. Clin. Investig. 2015, 125, 3338–3346.
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of tumors by the innate immune system and natural killer cells. Adv. Immunol. 2014, 122, 91–128.
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284.
- Clemente, C.G.; Mihm, M.C., Jr.; Bufalino, R.; Zurrida, S.; Collini, P.; Cascinelli, N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1996, 77, 1303–1310.
- Oldford, S.A.; Robb, J.D.; Codner, D.; Gadag, V.; Watson, P.H.; Drover, S. Tumor cell expression of HLA-DM associates with a Th1 profile and predicts improved survival in breast carcinoma patients. Int. Immunol. 2006, 18, 1591–1602.
- Kitamura, T.; Qian, B.-Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86.
- Cendrowicz, E.; Sas, Z.; Bremer, E.; Rygiel, T.P. The role of macrophages in cancer development and therapy. Cancers 2021, 13, 1946.
- Wang, M.; Huang, Y.K.; Kong, J.C.; Sun, Y.; Tantalo, D.G.; Yeang, H.X.A.; Ying, L.; Yan, F.; Xu, D.; Halse, H. High-dimensional analyses reveal a distinct role of T-cell subsets in the immune microenvironment of gastric cancer. Clin. Transl. Immunol. 2020, 9, e1127.
- Halse, H.; Colebatch, A.; Petrone, P.; Henderson, M.; Mills, J.; Snow, H.; Westwood, J.; Sandhu, S.; Raleigh, J.; Behren, A. Multiplex immunohistochemistry accurately defines the immune context of metastatic melanoma. Sci. Rep. 2018, 8, 11158.
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neuro-Oncol. Adv. 2023, 5, vdad009.
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell dysfunction in glioblastoma: Applying a new framework. Clin. Cancer Res. 2018, 24, 3792–3802.
- Miller, M.A.; Sullivan, R.J.; Lauffenburger, D.A. Molecular pathways: Receptor ectodomain shedding in treatment, resistance, and monitoring of cancer. Clin. Cancer Res. 2017, 23, 623–629.
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67.
- Wang, G.; Wang, J.; Niu, C.; Zhao, Y.; Wu, P. Neutrophils: New Critical Regulators of Glioma. Front. Immunol. 2022, 13, 927233.
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61.
- Lin, H.-C.; Song, T.-Y.; Hu, M.-L. S-Adenosylhomocysteine promotes the invasion of C6 glioma cells via increased secretion of matrix metalloproteinase-2 in murine microglial BV2 cells. Toxicol. Sci. 2009, 112, 322–330.
- Markovic, D.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kälin, R.; Van Rooijen, N. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535.
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527.
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949.
- Wang, H.; Zhou, H.; Xu, J.; Lu, Y.; Ji, X.; Yao, Y.; Chao, H.; Zhang, J.; Zhang, X.; Yao, S.; et al. Different T-cell subsets in glioblastoma multiforme and targeted immunotherapy. Cancer Lett. 2021, 496, 134–143.
- Alban, T.J.; Bayik, D.; Otvos, B.; Rabljenovic, A.; Leng, L.; Jia-Shiun, L.; Roversi, G.; Lauko, A.; Momin, A.A.; Mohammadi, A.M.; et al. Glioblastoma Myeloid-Derived Suppressor Cell Subsets Express Differential Macrophage Migration Inhibitory Factor Receptor Profiles That Can Be Targeted to Reduce Immune Suppression. Front. Immunol. 2020, 11, 1191.
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115.
- Mi, Y.; Guo, N.; Luan, J.; Cheng, J.; Hu, Z.; Jiang, P.; Jin, W.; Gao, X. The Emerging Role of Myeloid-Derived Suppressor Cells in the Glioma Immune Suppressive Microenvironment. Front. Immunol. 2020, 11, 737.
- Arce-Sillas, A.; Álvarez-Luquín, D.D.; Tamaya-Domínguez, B.; Gomez-Fuentes, S.; Trejo-García, A.; Melo-Salas, M.; Cárdenas, G.; Rodríguez-Ramírez, J.; Adalid-Peralta, L. Regulatory T Cells: Molecular Actions on Effector Cells in Immune Regulation. J. Immunol. Res. 2016, 2016, 1720827.
- Hoeppli, R.E.; Wu, D.; Cook, L.; Levings, M.K. The environment of regulatory T cell biology: Cytokines, metabolites, and the microbiome. Front. Immunol. 2015, 6, 61.
- Schmidt, A.; Oberle, N.; Krammer, P. Molecular Mechanisms of Treg-Mediated T Cell Suppression. Front. Immunol. 2012, 3, 51.
- Mammoto, T.; Jiang, A.; Jiang, E.; Panigrahy, D.; Kieran, M.W.; Mammoto, A. Role of Collagen Matrix in Tumor Angiogenesis and Glioblastoma Multiforme Progression. Am. J. Pathol. 2013, 183, 1293–1305.
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160.
- Walma, D.A.C.; Yamada, K.M. The extracellular matrix in development. Development 2020, 147, dev175596.
- Pietras, A.; Katz, A.M.; Ekström, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-CD44 Signaling in the Glioma Perivascular Niche Enhances Cancer Stem Cell Phenotypes and Promotes Aggressive Tumor Growth. Cell Stem Cell 2014, 14, 357–369.
- Chen, J.E.; Pedron, S.; Shyu, P.; Hu, Y.; Sarkaria, J.N.; Harley, B.A.C. Influence of Hyaluronic Acid Transitions in Tumor Microenvironment on Glioblastoma Malignancy and Invasive Behavior. Front. Mater. 2018, 5, 39.
- Lathia, J.D.; Li, M.; Hall, P.E.; Gallagher, J.; Hale, J.S.; Wu, Q.; Venere, M.; Levy, E.; Rani, M.R.; Huang, P.; et al. Laminin alpha 2 enables glioblastoma stem cell growth. Ann. Neurol. 2012, 72, 766–778.
- Jensen, G.; Holloway, J.L.; Stabenfeldt, S.E. Hyaluronic Acid Biomaterials for Central Nervous System Regenerative Medicine. Cells 2020, 9, 2113.
- Safarians, G.; Sohrabi, A.; Solomon, I.; Xiao, W.; Bastola, S.; Rajput, B.W.; Epperson, M.; Rosenzweig, I.; Tamura, K.; Singer, B.; et al. Glioblastoma Spheroid Invasion through Soft, Brain-Like Matrices Depends on Hyaluronic Acid–CD44 Interactions. Adv. Healthc. Mater. 2023, 12, 2203143.
- Stern, R.; Asari, A.A.; Sugahara, K.N. Hyaluronan fragments: An information-rich system. Eur. J. Cell Biol. 2006, 85, 699–715.
- Viola, M.; Vigetti, D.; Karousou, E.; D’Angelo, M.L.; Caon, I.; Moretto, P.; De Luca, G.; Passi, A. Biology and biotechnology of hyaluronan. Glycoconj. J. 2015, 32, 93–103.
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell 2006, 126, 677–689.
- Yang, Y.; Zheng, H.; Zhan, Y.; Fan, S. An emerging tumor invasion mechanism about the collective cell migration. Am. J. Transl. Res. 2019, 11, 5301–5312.
- Vazquez, K.; Saraswathibhatla, A.; Notbohm, J. Effect of substrate stiffness on friction in collective cell migration. Sci. Rep. 2022, 12, 2474.
- Gonçalves, I.G.; Garcia-Aznar, J.M. Extracellular matrix density regulates the formation of tumour spheroids through cell migration. PLoS Comput. Biol. 2021, 17, e1008764.
- Munson, J.M.; Shieh, A.C. Interstitial fluid flow in cancer: Implications for disease progression and treatment. Cancer Manag. Res. 2014, 6, 317–328.
- Malik, A.A.; Wennberg, B.; Gerlee, P. The Impact of Elastic Deformations of the Extracellular Matrix on Cell Migration. Bull. Math. Biol. 2020, 82, 49.
- Choquet, D.; Felsenfeld, D.P.; Sheetz, M.P. Extracellular Matrix Rigidity Causes Strengthening of Integrin–Cytoskeleton Linkages. Cell 1997, 88, 39–48.
- Steward, A.J.; Kelly, D.J. Mechanical regulation of mesenchymal stem cell differentiation. J. Anat. 2015, 227, 717–731.
- Sreenivasappa, H.; Chaki, S.P.; Lim, S.-M.; Trzeciakowski, J.P.; Davidson, M.W.; Rivera, G.M.; Trache, A. Selective regulation of cytoskeletal tension and cell–matrix adhesion by RhoA and Src. Integr. Biol. 2014, 6, 743–754.
- Song, L.; Dong, G.; Guo, L.; Graves, D.T. The function of dendritic cells in modulating the host response. Mol. Oral. Microbiol. 2018, 33, 13–21.
- Swartz, M.A.; Lund, A.W. Lymphatic and interstitial flow in the tumour microenvironment: Linking mechanobiology with immunity. Nat. Rev. Cancer 2012, 12, 210–219.
- Wiig, H.; Tveit, E.; Hultborn, R.; Reed, R.K.; Weiss, L. Interstitial fluid pressure in DMBA-induced rat mammary tumours. Scand. J. Clin. Lab. Investig. 1982, 42, 159–164.
- Flessner, M.F.; Choi, J.; Credit, K.; Deverkadra, R.; Henderson, K. Resistance of Tumor Interstitial Pressure to the Penetration of Intraperitoneally Delivered Antibodies into Metastatic Ovarian Tumors. Clin. Cancer Res. 2005, 11, 3117–3125.
- Li, R.; Serrano, J.C.; Xing, H.; Lee, T.A.; Azizgolshani, H.; Zaman, M.; Kamm, R.D. Interstitial flow promotes macrophage polarization toward an M2 phenotype. Mol. Biol. Cell 2018, 29, 1927–1940.
- Ye, D.; Desai, J.; Shi, J.; Liu, S.-Y.M.; Shen, W.; Liu, T.; Shi, Y.; Wang, D.; Liang, L.; Yang, S.; et al. Co-enrichment of CD8-positive T cells and macrophages is associated with clinical benefit of tislelizumab in solid tumors. Biomark. Res. 2023, 11, 25.
- Horibe, K.; Hara, M.; Nakamura, H. M2-like macrophage infiltration and transforming growth factor-β secretion during socket healing process in mice. Arch. Oral Biol. 2021, 123, 105042.
- Pfeifer, C.R.; Xia, Y.; Zhu, K.; Liu, D.; Irianto, J.; García, V.M.M.; Millán, L.M.S.; Niese, B.; Harding, S.; Deviri, D.; et al. Constricted migration increases DNA damage and independently represses cell cycle. Mol. Biol. Cell 2018, 29, 1948–1962.
- Belyaeva, V.; Wachner, S.; Gyoergy, A.; Emtenani, S.; Gridchyn, I.; Akhmanova, M.; Linder, M.; Roblek, M.; Sibilia, M.; Siekhaus, D. Fos regulates macrophage infiltration against surrounding tissue resistance by a cortical actin-based mechanism in Drosophila. PLoS Biol. 2022, 20, e3001494.
- Fanfone, D.; Wu, Z.; Mammi, J.; Berthenet, K.; Neves, D.; Weber, K.; Halaburkova, A.; Virard, F.; Bunel, F.; Jamard, C.; et al. Confined migration promotes cancer metastasis through resistance to anoikis and increased invasiveness. eLife 2022, 11, e73150.
- Caló, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267.
- Chin, M.H.W.; Norman, M.D.A.; Gentleman, E.; Coppens, M.-O.; Day, R.M. A Hydrogel-Integrated Culture Device to Interrogate T Cell Activation with Physicochemical Cues. ACS Appl. Mater. Interfaces 2020, 12, 47355–47367.
- Zhang, J.; Zhao, R.; Li, B.; Farrukh, A.; Hoth, M.; Qu, B.; del Campo, A. Micropatterned soft hydrogels to study the interplay of receptors and forces in T cell activation. Acta Biomater. 2021, 119, 234–246.
- Jacobelli, J.; Bennett, F.C.; Pandurangi, P.; Tooley, A.J.; Krummel, M.F. Myosin-IIA and ICAM-1 Regulate the Interchange between Two Distinct Modes of T Cell Migration1. J. Immunol. 2009, 182, 2041–2050.
- Wolf, K.; Müller, R.; Borgmann, S.; Bröcker, E.-B.; Friedl, P. Amoeboid shape change and contact guidance: T-lymphocyte crawling through fibrillar collagen is independent of matrix remodeling by MMPs and other proteases. Blood 2003, 102, 3262–3269.
- Lämmermann, T.; Sixt, M. Mechanical modes of ‘amoeboid’ cell migration. Curr. Opin. Cell Biol. 2009, 21, 636–644.
- Loo, J.; Sicher, I.; Goff, A.; Kim, O.; Clary, N.; Alexeev, A.; Sulchek, T.; Zamarayeva, A.; Han, S.; Calero-Garcia, M. Microfluidic transfection of mRNA into human primary lymphocytes and hematopoietic stem and progenitor cells using ultra-fast physical deformations. Sci. Rep. 2021, 11, 21407.
- Kwon, C.; Chung, A.J. Highly efficient mRNA delivery with nonlinear microfluidic cell stretching for cellular engineering. Lab. A Chip 2023, 23, 1758–1767.
- Camargo, C.P.; Muhuri, A.K.; Alapan, Y.; Sestito, L.F.; Khosla, M.; Manspeaker, M.P.; Smith, A.S.; Paulos, C.M.; Thomas, S.N. Adhesion analysis via a tumor vasculature-like microfluidic device identifies CD8(+) T cells with enhanced tumor homing to improve cell therapy. Cell Rep. 2023, 42, 112175.
- Coniglio, S.; Miller, I.; Symons, M.; Segall, J.E. Coculture Assays to Study Macrophage and Microglia Stimulation of Glioblastoma Invasion. J. Vis. Exp. 2016, 116, e53990.
- Zhou, C.; Wang, C.; Xu, K.; Niu, Z.; Zou, S.; Zhang, D.; Qian, Z.; Liao, J.; Xie, J. Hydrogel platform with tunable stiffness based on magnetic nanoparticles cross-linked GelMA for cartilage regeneration and its intrinsic biomechanism. Bioact. Mater. 2023, 25, 615–628.
- Hickey, J.W.; Dong, Y.; Chung, J.W.; Salathe, S.F.; Pruitt, H.C.; Li, X.; Chang, C.; Fraser, A.K.; Bessell, C.A.; Ewald, A.J.; et al. Engineering an Artificial T-Cell Stimulating Matrix for Immunotherapy. Adv. Mater. 2019, 31, e1807359.
- Li, X.; Sun, Q.; Li, Q.; Kawazoe, N.; Chen, G. Functional Hydrogels With Tunable Structures and Properties for Tissue Engineering Applications. Front. Chem. 2018, 6, 499.
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69.
- Labanieh, L.; Majzner, R.G.; Mackall, C.L. Programming CAR-T cells to kill cancer. Nat. Biomed. Eng. 2018, 2, 377–391.
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355.
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982.
- Shin, D.S.; Ribas, A. The evolution of checkpoint blockade as a cancer therapy: What’s here, what’s next? Curr. Opin. Immunol. 2015, 33, 23–35.
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106.