Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jifeng Zhang and Version 2 by Mona Zou.
Mature vascular smooth muscle cells (VSMC) exhibit a remarkable degree of plasticity, a characteristic that has intrigued cardiovascular researchers for decades. It has become increasingly evident that the chromatin remodeler SWItch/Sucrose Non-Fermentable (SWI/SNF) complex plays a pivotal role in orchestrating chromatin conformation, which is critical for gene regulation.
- vascular smooth muscle cell
- SWI/SNF complex
- epigenetics
- cardiovascular disease
1. Roles of VSMCs from Physiology to Pathology
Like skeletal muscle cells and cardiomyocytes, the primary function of VSMCs is contraction. Under precise hormonal and neural control, VSMCs regulate blood distribution and blood pressure. To maintain the contractile phenotype, VSMC undergoes differentiation via the expression of a repertoire of contractile apparatus and regulators [1][10]. Myosin heavy chain 11 (MYH11), also known as smooth muscle myosin heavy chain (SMMHC), is responsible for encoding SMC-specific myosin. Different from myosins in other muscle tissues, MYH11 lacks intrinsic ATPase activity. Instead, its activity is triggered when Ser 19 on the regulatory myosin light chain becomes phosphorylated. This phosphorylation is precisely controlled by myosin light chain kinase (MYLK) and myosin light chain phosphatase (MLCP). Thin myofilaments are composed of α-actin, also known as α-smooth muscle actin (α-SMA), which is encoded by actin alpha 2 (ACTA2) [2][3][11,12]. Of note, variants of those genes have already been well-documented in patients with inherited aortic diseases [4][5][13,14]. This underscores the importance of fully functional contractile machinery in VSMCs for maintaining aortic health.
Another important role of VSMCs is to regulate vascular extracellular matrix (ECM) homeostasis through the production of components including elastin, collagen [6][15], and proteoglycans and regulation of ECM remodelers including matrix metalloproteinase (MMP) and tissue inhibitor of matrix metalloproteinases (TIMPs). These determine the vessel wall’s mechanical strength, compliance, and elastic recoil. As a result, ECM construction and maintenance of VSMC have emerged as central priorities in the field of bioengineering for blood vessel grafts [7][16].
Interestingly, the two major roles of VSMCs seem to be conflicting. During the initial embryonic development stage, VSMCs exhibit rapid proliferation [8][17], migrate, and actively secrete ECM components crucial for vasculogenesis. However, mature VSMCs lose those properties and become quiescent with a low proliferation rate and synthetic activity. In the mature aorta, VSMCs possess contractile apparatus and maintain mechanical strength. Nevertheless, during vascular injury, alterations in the environmental cues cause the loss of contractile markers, including MYH11 and α-SMA, coupled with increased proliferation, migration, and protein synthesis in VSMC. This type of dedifferentiated VSMC is essential for vascular repair. The transition between the contractile and synthetic states of VSMCs is termed “phenotypic switch”. It is thus logical to infer that the intrinsic plasticity of VSMCs is beneficial for adaption to complex environments and response to damage. However, substantial changes in diet and lifestyle may hijack this ability and disrupt vascular homeostasis.
2. Epigenetic and Transcriptional Regulation of VSMC Plasticity in Health and Diseases
Vascular smooth muscle cell plasticity in CVD, including atherosclerosis, has been well-documented [9][2]. Especially in the past decade, the emergence of new technologies, such as lineage tracing, single-cell RNA sequencing (scRNA-Seq), and spatial transcriptomics [10][11][12][13][18,19,20,21], has largely extended our knowledge of cell origin, characteristics, and transcriptomic profiles in the atherosclerotic lesions. Under pathological conditions, VSMCs transdifferentiate into various cell types such as foam cells [14][15][22,23], mesenchymal-stem-cell (MSC)-like cells [16][24], macrophage-like cells [17][18][25,26], adipocyte-like cells [19][27], osteochondrogenic cells [20][21][28,29], fibromyocytes [11][19]. Multiple reviews thoroughly discuss this phenomenon [22][23][24][25][26][27][5,30,31,32,33,34]. A dramatic shift in gene expression occurs during dedifferentiation or transdifferentiation. Such changes necessitate a sophisticated transcriptional regulation network in VSMCs to precisely control specific gene expression in response to environmental changes [27][28][29][34,35,36].
In differentiated VSMC, the expressions of contractile genes are governed by an array of transcription factors and coactivators [28][29][30][35,36,37] (Table 1; Figure 1 and Figure 2). In 1985, CC(A/T)6GG, also referred to as the CArG element, was identified in the promoter of the human cardiac actin gene [31][38]. Subsequent research has highlighted the crucial role of the CArG element in regulating muscle-specific genes [32][39]. Serum response factor (SRF) was then identified to bind with CArG elements and regulate downstream gene expression [33][34][7,40]. In addition, the discovery of myocardin [35][8] and its competition with Elk-1 for SRF interaction [36][41] revealed the delicate regulation of the VSMC phenotype.
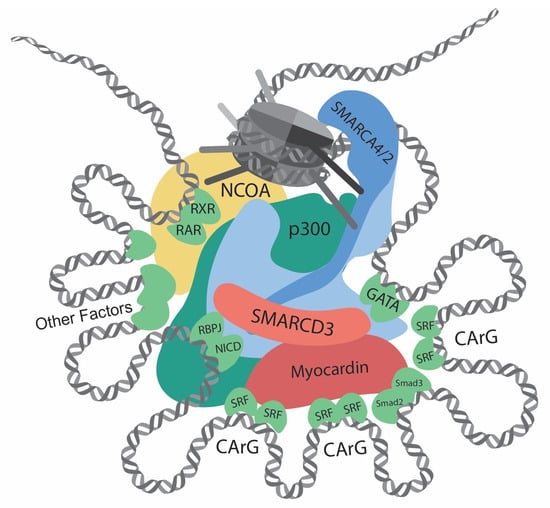
Figure 1. Transcriptional regulation at the CREs (cis-regulatory elements) of VSMC contractile genes. This diagram showcases the intricate network of interactions involving transcription regulators, such as chromatin remodelers, histone modifiers, transcription factors, and cofactors, at the CREs of the VSMC contractile genes. Chromatin remodelers that expose DNA regions by unwinding nucleosomes are central to the regulation. Several CArG elements, present at promoters and introns, bind to two SRFs and interact with myocardin. Within this region, there are binding sites for Smad2/3, NICD/RBPJ, and GATA. Retinoic acid signaling, crucial for VSMC differentiation, facilitates interactions between RAR/RXR and the SMARCD subunit of the SWI/SNF complexes. Other key contributors to VSMC differentiation include Prx1, Nkx3-2, PITX2, MEF2, and PIAS1. In addition, chromatin remodelers and transcription factors interact with histone modifiers, such as p300 and members of the NCOA family. Such complex interactions determine delicate and complicated regulation of contractile genes.
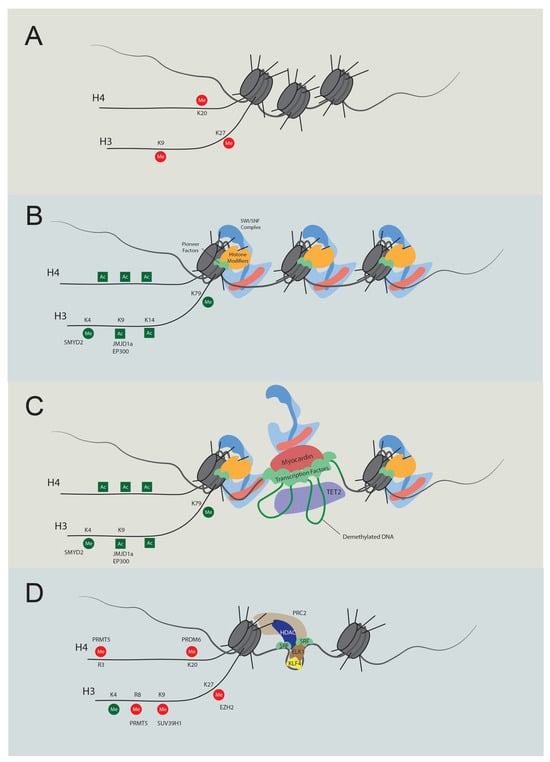
Figure 2. Temporal regulation of VSMC contractile gene expression. (A) Stem cell chromatin state: specific DNA regions are tightly wrapped into nucleosomes in stem cells. These regions are marked by repressive histone modifications, notably H4K20me3, H3K9me3, and H3K27me3, which play pivotal roles in maintaining gene silencing. (B) Activation of chromatin regions: during the transition to an active chromatin state, repressive histone marks are displaced, and active modifications emerge, such as H4Ac, H3K4Me2, H3K9Ac, H3K14Ac, and H3K79Me. Key players in this process include JMJD1a (H3K9me removal), SMYD2 (H3K4Me induction), and p300 (H3Ac and H4Ac induction). As histone acetylation weakens histone-DNA binding, pioneer factors can bind with nucleosomal DNA, subsequently recruiting either chromatin remodelers like the SWI/SNF complex or additional histone modifiers. (C) Transcription: the SWI/SNF complex introduces nucleosome-free regions, enabling TET2 to demethylate DNA. As a result, specific transcription factors bind to their respective elements. Subsequently, cofactors, including myocardin and CSRP, are recruited, promoting interactions between transcription regulators to promote transcription. (D) Dedifferentiation and transcriptional inactivation: in response to dedifferentiation signals like platelet-derived growth factor BB (PDGF-BB), the transcription of contractile genes was suppressed. In this situation, KLF4 binds to the G/C elements, while SRF partners with Elk1 rather than myocardin. Concurrently, HDACs and PRC are also recruited to deacetylate and induce methylation of H3K27. Additionally, PRDM6, PRMT5, and SUV39H1 mediate methylation at H4K20, H4R3 and H3R8, and H3K9, respectively. The loss of required transcription factors and DNA accessibility cease contractile gene transcription.
Table 1. Regulators of contractile gene transcription.
| Gene | Category | Function | Effect on Contractile Gene Transcription | Reference | ||
|---|---|---|---|---|---|---|
| SMARCA4 | Chromatin remodeler | Mediate chromatin accessibility | Dependent on interactors | [37][42[38],43] | ||
| SMARCD3 | Chromatin remodeler | Mediate chromatin accessibility | Promotion | [39][44] | ||
| SRF | Transcription factor | Bind at CArG elements | Dependent on interactors | [35][8] | ||
| Myocardin | Cofactor | Interact with SRF | Promotion | [35][36][40][8,41,45] | ||
| MRTFA/B | Cofactor | Interact with SRF | Promotion | [36][41][42][41,46,47] | ||
| Smad2/3 | Transcription factor | Bind DNA | Promotion | [43][48] | ||
| ARID1A/B | BAF250A/B | GATA4/6 | Transcription factor | Bind DNA | Promotion | [44][45][46][47][48][49,50, |
| ARID2 | BAF200 | 51 | , | 52 | ||
| PHF10 | BAF45A | |||||
| DPF1/2/3 | BAF45B/D/C | |||||
| BICRA/BICRAL | GLTSCR1/GLTSCR1L | |||||
| SMARCA4 | BRG1 | |||||
| SMARCA2 | BRM | |||||
| ACTL6A/B | BAF53A/B | |||||
| PBRM1 | BAF180 |
Recent advancements have significantly illuminated the structure of SWI/SNF complexes [106][107][108][109][113,114,115,116]. These findings further deepen our understanding of the chromatin remodeling mechanism of SWI/SNF complexes and the function of their subunits. According to Chen et al., SWI/SNF complexes can be divided into three modules: the motor module, actin-related protein (ARP) module, and substrate recruitment module (SRM) [110][117]. SRM can be further divided into nucleosome binding lobe (NBL), DNA binding lobe (DBL), and histone-tail binding lobe (HBL) [110][117]. The central motor module is the ATPase, SMARCA4 (BRG1), and SMARCA2 (BRM), which directly hydrolyze ATP to translocate DNA [111][118]. The ARP module comprises actin beta (ACTB), BCL7, and actin-like 6 (ACTL6). They serve the dual function of connecting ATPase with SRM and regulating the ATPase activity. Within the nucleosome binding lobe, SMARCB1, coupled with double PHD fingers (DPF) and SMARCC, attaches directly to the nucleosome acidic patch via the C-terminal domain (CTD) [112][119]. The DNA binding lobe interacts with extranucleosomal linker DNA and various factors. This lobe consists of several DNA binding domains like the HMG-box domain on SMARCE1 and polybromo 1 (PBRM1) [113][120], AT-rich interaction domain (ARID) on ARID1A/B, and ARID2 [114][121].
Of note, multiple interactors of SWI/SNF complexes exist to orchestrate gene transcription (some interactions validated by the GST pull-down assay are listed in Table 3). The SMARCD family, including SMARCD1, SMARCD2, and SMARCD3, are reported to mediate the interactions between the SWI/SNF complex and various factors, such as PPARG coactivator 1α (PGC1α) and CCAAT enhancer binding protein ε (CEBPε) [115][116][122,123]. Recently, Wolf et al. [117][124] fused TurboID [118][125] with SMARCD1 and revealed close associations of the SWI/SNF complex with various transcription factors and epigenetic machinery, including lysine-specific methyltransferase 2 (KMT2) family, nuclear receptor coactivator (NCOA) family and histone acetyltransferases (HATs). The SWI/SNF complex can also recognize histone modifications. The bromodomains within its subunit BRD7/9, SMARCA4, SMARCA2, and PBRM1 facilitate their binding to acetylated histones [119][126]. The chromodomains on the subunit SMARCC1 and SMARCC2 recognize methylated H3 [120][127]. Given their sophisticated structure and interactors, it is speculated that the whole complexes remodel the chromatin at precise locations and accurate time points.
Table 3. SWI/SNF subunit-interacting proteins validated by GST pull-down assay.
| Interactor | SWI/SNF Subunit | Reference | |||||
|---|---|---|---|---|---|---|---|
| AR | SMARCC1 | [121][128] | |||||
| CBP | SMARCA4 | [122][129] | |||||
| ERα | SMARCD1 | [123][130] | |||||
| ERα | SMARCD3 | [124][131] | |||||
| FOS | SMARCD1 | [125][132] | |||||
| JUN | SMARCD1 | [125][132] | |||||
| , | 53 | ] | |||||
| JUN | SMARCD3 | [124][131] | CSRP2 | Cofactor | Interact with SRF, GATA6 | Promotion | [49][50] |
| MYC | [ | 54 | SMARCA2 | [126][133],55] | |||
| NKX3-2 | Transcription factor | Bind DNA | Promotion | [47] | |||
| MYC | SMARCA4 | [126][133] | [ | 52] | |||
| Prx1 | Transcription factor | Bind DNA | Promotion | [ | |||
| MYC | SMARCB1 | [126] | 51 | ][56] | |||
| [ | 133 | ] | PITX2 | Transcription factor | Bind DNA | Promotion | [52][57] |
| MYC | SMARCE1 | [126][133] | PIAS1 | Transcription factor | Bind DNA | Promotion | [53 |
| Myocardin | SMARCD3 | [127][ | ] | [58] | |||
| 134 | ] | MEF2 | Transcription factor | Bind DNA | Promotion | [54][59] | |
| NCOA1 | ARID1 | [128][135] | Notch/RBPJ | Transcription factor | Bind DNA | Promotion | [55][56 |
| NCOA1 | ] | [ | SMARCC1 | [121][128]60,61] | |||
| KLF4 | Transcription factor | Bind G/C repressor element | Repression | [57][58][59][60][61][9,62,63,64,65] | |||
| Elk1 | Cofactor | Interact with SRF | Repression | [42][59][60][47,63,64] | |||
| p300 | Histone acetyltransferase | Increase histone acetylation | Promotion | [46][62][51[63],66,67] | |||
| HDAC | Histone deacetylase | Decrease Histone modification | Repression | [42][59][60][47,63,64] | |||
| SMYD2 | Histone lysine methyltransferase | Increase H3K4me1, H3K4me3 | Promotion | [64][68] | |||
| JMJD1A | Histone demethylase | Decrease H3K9me2 | Promotion | [65][69] | |||
| WDR5 | Cofactor | Increase H3K4me1, H3K4me3 | Promotion | [66][70] | |||
| PRDM6 | Histone lysine methyltransferase | Increase H4K20me2 | Repression | [67][68][71,72] | |||
| SUV39H1 | Histone lysine methyltransferase | Increase H3K9me3 | Repression | [69][73] | |||
| EZH2 | Histone lysine methyltransferase | Increase H3K27me3 | Repression | [70][71][74,75] | |||
| TET2 | Methylcytosine dioxygenase | DNA demethylation | Promotion | [72][73][76,77] | |||
| PRMT5 | Histone arginine methyltransferase | H3R8me2, H4R3me2 | Repression | [74][78] |
Aside from myocardin/SRF, other transcription factors also play essential roles in regulating contractile genes [28][35]. The roles of transforming growth factor-β (TGFβ) in VSMC differentiation have been documented [75][79]. In 1997, the TGFβ control element (TCE), proximal to two CArG elements, was discovered in the promoter of ACTA2 [76][80]. TGFβ increases contractile gene expression by facilitating the binding of SRF to the CArG elements, possibly through interactions between Smad3 and p300 [77][81]. Subsequent research on the synergetic function and direct interaction of Smad3 and myocardin has further elucidates the roles of TGFβ signaling in VSMC differentiation [43][48]. Another transcription factor, GATA binding protein 6 (GATA6), was found to be highly expressed in the VSMCs during development [78][82], and it was shown to protect against injury-induced VSMC phenotypic switch [45][50]. Further study has revealed that GATA6, NK3 homeobox 2 (NKX3-2), and SRF form a triad complex to regulate contractile gene expressions [47][52].
As we delve deeper into the research on the transcriptional regulation of contractile genes, epigenetic regulation gains increasing interest [79][80][87,88]. Structural investigations revealed that SRF does not bind to nucleosomal DNA [81][82][83][89,90,91], implying its exclusive binding to open DNA regions. Meanwhile, several factors affect the binding activity of SRF to the CArG elements [76][80][84][80,86,88]. These findings shed light on the significance of chromatin conformation in the transcriptional regulation of contractile genes. McDonald et al. discovered that H3K4me2, H3K79me2, H3K9Ac, and H4Ac are enriched in the CArG elements in the SMC but not in the non-SMC [84][86]. In addition, H3K4me2 tethers with SRF and myocardin, and this modification persists even after VSMC dedifferentiation [85][92]. Loss of H3K4me2 at the CArG elements causes a decrease in contractile gene expression through the reduced TET2 (ten-eleven translocation-2)-mediated DNA demethylation [73][86][77,93]. In contrast, in embryonic stem cells or other non-SMCs, H3K9me3 and H3K27me3 govern the repression of contractile gene expression [83][91]. Lysine demethylase 3A (KDM3A), previously known as JMJD1a, interacts with myocardin and demethylates H3K9me3, thus promoting the expression of contractile genes [65][69]. In addition, numerous histone modifiers have been identified to regulate contractile genes, including PR/SET domain 6 (PRDM6), SET and MYND domain containing 2 (SMYD2), SUV39H1, polycomb repressive complexes 2 (PRC2), and protein arginine methyltransferase 5 (PRMT5) (Table 1; Figure 1 and Figure 2) [64][67][68][69][70][71][74][68,71,72,73,74,75,78]. Although previous studies elucidated the intricate regulation of transcription and histone modification, the specific roles of chromatin remodelers in directly executing chromatin transformations require further investigation.
3. Chromatin Remodeling and SWI/SNF Complexes
In eukaryotic cells, DNA wraps around histones, forming the basic structural unit: a nucleosome [87][94]. In a rigid and delicate manner, DNA accessibility is highly regulated for complex activities, including replication, repair, and transcription. Chromatin remodeling involves altering interactions between histones and DNA, including assembly and disorganization of nucleosomes. Transcription requires DNA free from histones to interact with proteins such as polymerase II and transcription factors. Studies have shown that chromatin remodelers are essential in controlling pluripotency, cell fate, and differentiation [88][89][90][95,96,97]. Four prominent remodeler families have spiked the most study interest to date, including SWI/SNF, Imitation switch (ISWI), chromodomain helicase DNA-binding (CHD), and INOsitol requiring 80/SWI2/SNF2-Related 1 (INO80/SWR1) [91][92][98,99]. This review focuses on the SWI/SNF complexes.
The SWI/SNF chromatin remodeling complexes were first discovered in Saccharomyces cerevisiae. SWI stands for ‘switch’ as the relevant genes regulate the HO gene, which is crucial for mating type switching [93][100]. Similarly, SNF denotes ‘sucrose nonfermenting’ since these genes control the SUC2 gene responsible for sucrose catabolism [94][101]. Subsequent research has found that some genes from these two screenings overlap, forming a complex that regulates chromatin structure [95][102]. SWI/SNF complexes use energy from ATP hydrolysis to mediate nucleosome sliding or ejection [96][103]. The mammalian SWI/SNF complex was purified in 1996 [97][104]. In mammals, either Brg1 or Brm serves as the ATPase of the complex; the SWI/SNF complex is also named the BRG1/BRM-associated factor (BAF). The initially isolated complex was termed canonical BAF (cBAF) [97][104]. Subsequent research has identified two more types of SWI/SNF assemblies: polybromo-associated BAF (PBAF) [98][105] and non-canonical BAF (ncBAF) [99][100][101][106,107,108]. These complexes are assembled by 9–16 subunits, and some subunits consist of several paralogs (Figure 3 and Table 2) [102][109]. Each subunit conveys unique functions for chromatin remodeling [103][110]. More than 1000 unique combinations of SWI/SNF subunits form diverse complexes with varied functions [104][105][111,112].
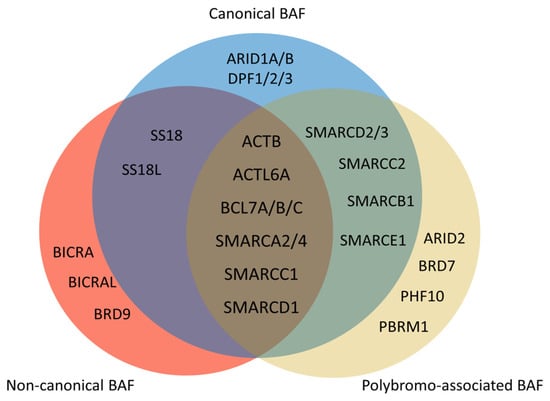
Figure 3. Composition of mammalian SWI/SNF complexes. The mammalian SWI/SNF chromatin remodeling complexes exist in three distinct types of assemblies: canonical BAF (cBAF), non-canonical BAF (ncBAF), and polybromo-associated BAF (PBAF). All three share several common subunits, such as the ATPase SMARCA2/4 and the ARP module (which includes ACTB, ACTL6A, and BCL7A/B/C). There are several differences between three types of assemblies. ARID1A/B and DPF1/2/3 only exist in cBAF. There are ARID2, BRD7, PHF10, and PBRM1 but no SS18/SS18L in PBAF. Non-canonical BAF possesses BICRA, BICRAL, and BRD9, and lacks SMARCC2, SMARCD2/3, SMARCB1, and SMARCE1.
Table 2. SWI/SNF complex subunit HUGO name and common name.
| HUGO Name | Common Name | |
|---|---|---|
| SMARCC1 | BAF155, SRG3 | |
| SMARCC2 | BAF170 | |
| SMARCD1/2/3 | BAF60A/B/C | |
| SMARCB1 | BAF47, INI1 | |
| SMARCE1 | BAF57 | |
| NCOA1 | ||
| SMARCE1 | ||
| [ | ||
| 128 | ||
| ] | ||
| [ | 129 | ][135,136] |
| Nkx2-5 | SMARCD3 | [127][134] |
| NR3C1/GR | SMARCD1 | [123][130] |
| NR3C1/GR | SMARCE1 | [123][130] |
| PCG1α | SMARCD1 | [116][123] |
| PPARγ | SMARCD1 | [116][123] |
| PPARγ | SMARCD3 | [124][131] |
| PRMT5 | SMARCB1 | [126][133] |
| PRMT5 | SMARCE1 | [126][133] |
| RAR | SMARCD3 | [128][135] |
| RBP-J | SMARCD3 | [130][137] |
| RORα | SMARCD3 | [124][131] |
| RXR | SMARCD3 | [124][128][131,135] |
| SREBP1α | SMARCD3 | [124][131] |
| Tbx5 | SMARCD3 | [127][130][134,137] |
4. SWI/SNF Complex in Cardiovascular Development
Coffin–Siris syndromes (CSSs) represent congenital disorders predominantly linked to mutations in the subunits of SWI/SNF complexes and are characterized by mental retardation [131][138]. ARID1B is responsible for CSS1, with ARID1A for CSS2, SMARCB1 for CSS3, SMARCA4 for CSS4, SMARCE1 for CSS5, ARID2 for CSS6, DPF2 for CSS7, SMARCC2 for CSS8, SMARCD1 for CSS11, and BICRA for CSS12. Recently, more phenotypes in the cardiovascular system have been noticed in CSS patients [132][139]. Among fetuses, 67% of patients present cardiac anomalies, and 53% of patients present vascular anomalies [133][140]. For instance, one case study has reported that one fetus with SMARCC2 deficiency was diagnosed with tetralogy of Fallot, a rare congenital disease caused by a combination of four heart defects [134][141]. Considering the lethality of cardiovascular anomality and the higher possibility of termination due to poor prognosis, the actual incidence of cardiovascular complications in CSS might be underestimated.
Over decades of research on the mammalian SWI/SNF complex, numerous animal models have been developed. In the mouse model, SMARCA4 null embryo dies at day three due to the arrest of differentiation [135][142], while SMARCA2 knockout mice are viable [136][143]. This indicates that the roles of SMARCA4 and SMARCA2 differ in development. As an essential subunit in the SWI/SNF complex, DPF3 is highly expressed in the heart and skeletal muscle and serves as a histone modification reader. DPF3 possesses 2 plant homeodomains (PHDs), which facilitate its binding with acetylated H3 and H4, and H3K4me1/2. Knockdown dpf3 in the zebrafish leads to irregular cardiac morphology and muscular fiber disarray [137][144]. Constitutive knockout of BIRCA (BRD4 interacting chromatin remodeling complex associated protein) (also known as GLTSCR1 (glioma tumor suppressor candidate region gene 1)) results in embryonic lethality and cardiac defects, including ventricular septal defect, double outlet right ventricle and a thinner ventricular wall [138][145]. These results indicate the indispensable roles of the SWI/SNF complex in the development of the cardiovascular system.