Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Wendy Huang and Version 1 by Ana Vuletić.
Histone deacetylase 6 (HDAC6), by deacetylation of multiple substrates and association with interacting proteins, regulates many physiological processes that are involved in cancer development and invasiveness such as cell proliferation, apoptosis, motility, epithelial to mesenchymal transition, and angiogenesis. Due to its ability to remove misfolded proteins, induce autophagy, and regulate unfolded protein response, HDAC6 plays a protective role in responses to stress and enables tumor cell survival.
- HDAC6
- damaged proteins
- degradation
- autophagy
- molecular chaperone
- apoptosis
- NLRP3 inflammasome
- immune responses
1. Introduction
HDAC6 has gained a lot of attention since its discovery in 1999 [20][1]. It consists of 1215 amino acids and possesses five functional domains (Figure 1). Starting from its N- to the C- terminus, HDAC6 comprises the following regions: a nuclear localization sequence (NLS) that is rich in arginine (Arg) and Lys, a nuclear export sequence (NES) that is rich in leucine (Leu), two catalytic deacetylase domains (CD1 and CD2), the cytoplasmic anchoring serin (Ser) glutamine (Glu)-containing tetrapeptide (SE14), and a ubiquitin-binding zinc finger motif domain (ZnF-UBP). NLS and NES together control the trafficking of HDAC6 between the nucleus and the cytoplasm, while SE14 is responsible for the intracellular retention of HDAC6. HDAC6 also contains a dynein motor-binding sequence (DMBS) between the CD1 and CD2 catalytic domains [17,20,21,22][1][2][3][4].
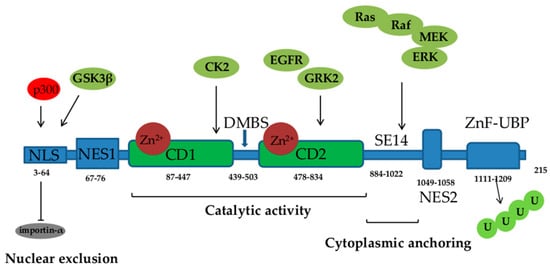
Figure 1. Functional domains of histone deacetylase (HDAC) 6: NLS—nuclear localization sequence, NES—nuclear export sequence; two catalytic domains (CD1 and CD2), DMBS—dynein motor-binding sequence, ZnF-UBP—zinc finger ubiquitin-binding domain, U—ubiquitin. Acetylation of NLS by p300 inhibits HDAC6 interaction with importin-α. Ras/Raf/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK), epidermal growth factor receptor (EGFR), G protein-coupled receptor kinase (GRK) 2, casein kinase (CK) 2, and glycogen synthase kinase (GSK) 3β signaling phosphorylate and activate HDAC6.
With the two catalytic domains, HDAC6 is a unique class IIb HDAC, responsible for the deacetylation of a number of non-histone substrates involved in the regulation of crucial physiological processes, including cell proliferation, survival, apoptosis, autophagy, motility, intracellular transport, and stress responses. HDAC6 regulates the deacetylation of multiple cytoplasmic substrates and affects their activity, cellular location, and protein–protein interactions. The deacetylase activity of both catalytic domains is Zn2+ dependent. The CD1 catalytic domain mostly deacetylates C-terminal acetyl-Lys residues and cannot independently exert catalytic activity but needs CD2 assistance. Aside from its deacetylase activity, HDAC6, through the catalytic CD1 domain, exhibits E3 ubiquitin ligase activity [21][3]. Proteins in the cytoplasm that are identified as substrates for deacetylation by HDAC6 include α-tubulin, cortactin, heat shock protein (Hsp) 90, heat shock transcription factor-1 (HSF-1), Ku70, p53, peroxiredoxins, signal transducer and activator of transcription (STAT) 3, forkhead box protein O1 (FOXO1), and β-Catenin [17,18,23,24,25][2][5][6][7][8] (Table 1).
Table 1.
HDAC6 substrates and physiological functions of HDAC6-mediated deacetylation.
| Protein | Localization | Function | Reference |
|---|---|---|---|
| α-tubulin | Cytoplasm | Microtubule disassembly, increases cell motility | [26,27,28,29][9][10][11][12] |
| Cortactin | Cytoplasm | Actin polymerization and branching, increases cell motility | [30][13] |
| TFEB | Cytoplasm | Autophagy | [31][14] |
| FOXO1 | Cytoplasm | Autophagy | [32][15] |
| Hsp90 | Cytoplasm | Degradation of misfolded proteins | [33][16] |
| GRP78 | Cytoplasm and nucleus | ER stress regulation, tumor progression via secretion of exosomes | [34][17] |
| NF-κB | Nucleus | Transcription of genes for NLRP3, pro-IL-1β, pro-IL-18, inflammasome activity | [35][18] |
| P53 | Cytoplasm | Cell cycle progression, inhibition of apoptosis, induced autophagy via upregulation of Beclin-1 | [36,37,38][19][20][21] |
| Ku70 | Cytoplasm | Suppression of apoptosis | [25,39][8][22] |
| Survivin | Nucleus | Suppression of apoptosis | [40,41][23][24] |
| Peroxidins I and II | Cytoplasm and nucleus | Antioxidant activity | [42][25] |
| Smad3 | Cytoplasm | Downregulation of E-cadherin expression, EMT | [43,44][26][27] |
| β-catenin | Cytoplasm | Translocation into nucleus and tumor cell invasion | [45][28] |
| STAT3 | Cytoplasm | Activation of JAK/STAT3 signaling and inflammatory responses | [46][29] |
| TAK1 | Cytoplasm | Activation of ADAM17 MMP enhances sIL-6R release and M2 macrophage differentiation | [47][30] |
| ERK1 | Cytoplasm | Activation of ERK1, proliferation, survival, and increased cell motility | [48][31] |
| AKT | Cytoplasm | Activation of AKT pathway, cell migration | [49][32] |
TFEB—transcription factor EB, Hsp—heat shock protein 90, GRP—glucose-regulated protein 78, ER—endoplasmic reticulum, NLRP3—NLR family pyrin domain-containing 3, EMT—epithelial–mesenchymal transition, JAK—Janus kinase, STAT—signal transducer and activator of transcription 3, ADAM—A disintegrin and metalloproteinase-17, MMP—matrix metalloproteinase, sIL-6R—soluble IL-6 receptor, TAK—Transforming Growth Factor-β-activated kinase 1, STAT—signal transducer and activator of transcription 3, ERK—extracellular signal-regulated kinase 1.
2. Cytoskeleton Organization
HDAC6 affects cytoskeletal structure and dynamics by modifying microtubule and actin organization and thereby is involved in the maintenance of cellular shape, cell division, cell migration, intracellular transport, and angiogenesis. The cytoskeletal component, α-tubulin, is acetylated at Lys40 by α-tubulin acetyltransferase (αTAT), which enables tubulin polymerization and formation of microtubules, while its deacetylation by HDAC6 promotes microtubule disassembly [27,28][10][11]. The overexpression of HDAC6 is associated with tubulin deacetylation, proclivity to chemotactic movement, and increased cell motility [29][12]. Furthermore, HDAC6 deacetylates another cytoskeletal protein cortactin that is present in areas of dynamic actin assembly such as at the leading edge of migrating cells. Deacetylated cortactin subsequently binds to F-actin through the small GTPase Rac1 and the actin nucleating complex Arp2/3, which enhances actin polymerization and branching, leading to increased cell motility. Cortactin is often overexpressed in tumors [65][33], including colorectal canncer (CRC), where it promotes malignant cell proliferation by activating the EGFR–MAPK pathway. Its expression was reported to correlate with the metastatic potential of a tumor [30,66][13][34]. The acetylation of cortactin prevents its translocation to the cell periphery, blocks its association with F-actin, and impairs cell motility, while its deacetylation by HDAC6 increases tumor cell motility and hence their invasiveness [26,30][9][13].3. Degradation of Damaged Proteins
HDAC6 mediates the clearance of misfolded and damaged proteins that are often accumulated in malignant cells. Although the majority of defective proteins are labeled with ubiquitin chains and degraded by the ubiquitin–proteasome (UP) pathway, if the proteasome system is overwhelmed or if protein aggregates are unsuitable for UP degradation, they are degraded in autophagosomes. Cancer cells are characterized by extensive autophagic activity that modulates the recycling of cellular proteins, regulates cellular homeostasis and energy metabolism, enables uncontrollable proliferation, protects cells from stress imposed by accumulated damaged proteins, and promotes the survival of malignant cells [67][35]. HDAC6, through its UBP domain, simultaneously interacts with polyubiquitinated proteins while its DMBS interacts with the p150glued component of the dynein motor complex [27[10][36],54] (Table 2), and in this manner, it facilitates the transport of aggregated or misfolded proteins toward the microtubule-organizing center (MTOC) [68][37]. Furthermore, HDAC6 mediates the formation of aggresomes around the misfolded protein by deacetylating cortactin, which subsequently interacts with F-actin, triggering actin polymerization and fusion of aggresomes with autophagosomes and lysosomes to lyse proteins [69][38]. The ability of HDAC6 to mediate the formation and removal of aggresomes is regulated by casein kinase (CK) 2, which phosphorylates HDAC6 and increases its deacetylase activity [70][39]. HDAC6 coordinates the UP system and autophagy maintaining them in a complementary relationship by interacting with the scaffolding protein sequestosome 1 (SQSTM1)/P62. P62 functions as a ubiquitin-recognizing receptor that binds ubiquitinated proteins and encapsulates them into aggresomes, where they fuse with lysosomes and become degraded [71][40]. Furthermore, the E3-ubiquitin ligase TRIM50 promotes the recruitment and aggregation of polyubiquitinated (poly-U) proteins into aggresomes by facilitating the interactions of HDAC6 and p62 [53][41]. In this way, HDAC6 improves the efficiency and selectivity of autophagic degradation [72,73][42][43]. While TRIM50 promotes the clearance of ubiquitinated proteins in aggresomes, an HDAC6-interacting chaperone, valosin-containing protein p97/(VCP) ATPase, induces the dissociation of HDAC6 and poly-U proteins and protein delivery to proteasomes. In this sense, the excess of p97/VCP favors protein degradation in proteasomes [55][44]. It has been established that p62 and ubiquitin are highly expressed in colon carcinoma and that high ubiquitin expression has an impact on the number of lymph node metastases in patients with CRC [74][45]. Although, in some tumors, the expression of cytoplasmic p62 was negatively associated with patients’ survival, in CRC, a favorable prognostic significance of cytoplasmic p62 was found in the mutated K-RAS but not in the wild-type (wt) K-RAS subgroup of patients [75][46].4. Autophagy
Aside from the formation of aggresomes and the transportation and degradation of autophagosomes, HDAC6 regulates autophagy by deacetylating autophagy-related transcription factors and proteins [76][47]. In this sense, HDAC6 deacetylates transcription factor EB (TFEB) [31][14] and forkhead box O1 (FOXO1) [32][15] to decrease their activity and to inhibit autophagy. TFEB and FOXO transcription factors are inactivated by phosphorylation [77][48]. HDAC6 inhibition promotes the acetylation of TFEB, which then enhances the expression of the autophagy-related protein Beclin-1 [78][49]. However, there are opposing data on the effect of HDAC6 on Beclin-1 expression obtained on liver cancer cells, showing that HDAC6 overexpression activated c-Jun NH2-terminal kinase (JNK) and increased the phosphorylation of c-Jun, which induced Beclin-1-dependent autophagy [79,80][50][51]. The acetylated FOXO1 transcription factor is also required for T-cell differentiation into regulatory T (Treg) cells expressing Foxp3 transcription factors [81][52]. It was reported that the pharmacological inhibition of HDAC6 enhanced the transcriptional activity of acetylated FOXO1 and facilitated the autophagy process [32,80][15][51]. Furthermore, HDAC6 regulates the activity of an autophagy-related protein, the microtubule-associated protein 1 light chain 3 (LC3). LC3-I forms a conjugate with phosphatidylethanolamine (PE) and becomes LC3-II, which is then transported by HDAC6 to the MTOC to promote autophagosome formation [80,82][51][53]. It was reported that the deacetylation of LC3-II by HDAC6 promotes its translocation to the cytoplasm and autophagy, thereby inducing the survival of nutrient-deprived tumor cells [80,83][51][54]. This suggests that HDAC6 functions both as a scaffold protein and as a deacetylase in the regulation of LC3 that promotes autophagy [80][51]. Cytoskeletal modifications by HDAC6 are relevant for the progression of autophagy. While the actin remodeling induced by cortactin deacetylation by HDAC6 is essential for the fusion of autophagosomes and lysosomes [69][38], the deacetylation of α-tubulin and microtubule disassembly inhibits their fusion with lysosomes [84][55]. It was reported that the expression of nuclear Beclin-1 and LC3 in patients with CRC harboring K-RAS mutations is associated with shorter overall survival [75][46]. Moreover, according to one study, the activation of the KRAS/BRAF/phosphoinositide 3-kinase (PI3K) oncogenic pathway by KRAS and BRAFV600E mutations induces the expression of the key autophagic markers lLC3 and Beclin-1 in CRC cells, thus promoting autophagy [85][56].5. Regulation of Molecular Chaperones and Other Stress-Related Proteins
HDAC6 affects protein degradation in proteasomes by the deacetylation of the Hsp90 molecular chaperone that has a primary physiological ability to stabilize protein tertiary structures, regulate transportation, and prevent protein degradation in proteasomes, thus enabling the biological functions of its client protein [56][57]. Multiple client proteins are regulated by Hsp90, such as steroid hormone receptors, growth factor receptors (epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGFR)), molecules engaged in the regulation of the cell cycle and apoptotic pathways, transcription factors such as hypoxia-inducible factor 1-α (HIF-1α), signaling molecule RAF Ser/Thr protein kinase, etc. The acetylation of Hsp90 impairs its chaperone activity and therefore induces degradation of the client protein. Simultaneously, the stability of HDAC6 is modulated by Hsp90 [33][16], while the degradation of HDAC6 is regulated by cullin 3SPOP ubiquitin E3 ligase that has been shown to promote HDAC6 polyubiquitination and degradation in proteasomes in multiple cancer cell lines, including CRC [86][58]. HDAC6 also regulates the function of Hsp90 through the ZnF-UBP domain. HDAC6 senses ubiquitinated protein aggregates but also indirectly induces the expression of molecular chaperones, as it can activate p97/VCP that through its enzymatic activity induces the release of HSF1 from the Hsp90/HSF1 complex [54,55][36][44]. The released HSF1 induces the expression of genes for molecular chaperones Hsp70 and Hsp90 [57][59], which is followed by the release of HDAC6 from the complex and its binding to ubiquitinated proteins [56,67][35][57]. In CRC, the expression of Hsp90 in tumor tissue was inversely associated with survival outcomes and could represent a potential unfavorable prognostic factor for CRC patients [87][60]. As HDAC6 protects tumor cells from cytotoxic effects caused by defective and misfolded proteins, the inhibition of HDAC6 and Hsp90 may have therapeutic potential in cancer. In this sense, inhibition of Hsp90 has shown an antitumor effect in animal models of CRC and in CRC cell lines as it caused the depletion of B-RAF and K-RAS, which are major oncogenic drivers in CRC associated with poor disease prognosis [88][61]. HDAC6 plays an active role in the response to environmental stress imposed by newly synthesized secretory proteins in the endoplasmic reticulum (ER) in order to eliminate misfolded proteins before protein aggregation becomes lethal for the cell [89][62]. In this sense, HDAC6 targets the 78 kDa glucose-regulated protein (GRP78) and Hsp70, the molecular chaperones that are involved in the unfolded protein response (UPR) that implies the regulation of proper folding, conformational maturation, assembly of proteins in ER, and control of the overall quality of proteins. Aside from its role in the UPR, GRP78 displays antiapoptotic properties, promotes tumor proliferation, survival, and metastasis, and confers resistance to chemotherapy. The level of GRP78 was inversely associated with the sensitivity of CRC cells to alkylating agents, including cisplatin and 5-FU [90][63]. In solid tumors, hypoxic conditions, acidosis, and glucose deficiency induce GRP78 expression [91][64]. It has been shown that colon cancer cells secrete GRP78 via exosomes and that this process is dependent on the activity of HDAC6 [34][17], which is often overexpressed in CRC [19][65]. HDAC6 inhibition increases GRP78 acetylation. Subsequently, the acetylated GRP78 dissociates from HDAC6 and then binds to VPS34, a class III PI3K, thus preventing the sorting of GRP78 into multivesicular bodies and GRP78 release that induces its aggregation in the ER that further inhibits tumor growth [34][17]. Moreover, by inhibiting the release of exosomes containing GRP78 from cancer cells, HDAC6 inhibition also inhibits angiogenesis as GRP78 is involved in blood vessel formation in growing tumors through the activation of HIF-1α and VEGF/VEGFR, as well as the PI3K/AKT, ERK, and FAK signaling pathways [92][66]. HDAC6 has an important role in redox regulation in response to cellular stress. HDAC6 deacetylates the redox-regulatory antioxidant enzymes peroxiredoxin (Prx) I and Prx II [42][25]. Prxs are often present at high levels in cancer and neurodegenerative disorders and play a protective role against oxidative damage. The acetylation of Prx increases its reductase activity, thus, HDAC6 and Prx may be considered as therapeutic targets for modulating intracellular redox status in cancer [22][4].6. Apoptosis
HDAC6 affects cell cycle progression and apoptosis by modulating the activity of multiple proteins, including p53. Upon DNA damage, p53 is activated by the kinases ATM, ATR, Chk1, and Chk2. This leads to the disruption of the interaction between p53 and mouse double minute 2 homolog (MDM2), which is an E3 ubiquitin-protein ligase [93,94][67][68]. p53 can be acetylated by the acetyltransferases CBP and p300, which prevent its ubiquitination and enhance its stability and transcriptional activity toward the expression of proapoptotic proteins Bax and Puma [36,37,95][19][20][69]. Moreover, acetylated p53 releases the apoptotic molecule Bax from the nuclear p53/Bax complex, which is then translocated to the mitochondria to induce cytochrome C release and apoptosis [94,95][68][69]. HDAC6 has been found to deacetylate p53 and repress its function as a tumor suppressor [36,37,38][19][20][21]. It was reported that HDAC6 inhibition increases the acetylation of p53 in tumor cells, which leads to upregulated expression of genes related to cell cycle control and apoptosis, including p21 cyclin-dependent kinase (CDK) inhibitor, which can be induced with HDAC inhibition in p53-dependent and -independent ways [37,96][20][70]. Moreover, acetylated cytoplasmic p53 inhibits autophagy by inducing Beclin-1 degradation via the ubiquitin-specific peptidase USP10 and by inhibiting the mTOR (mechanistic target of rapamycin complex) pathway [38][21]. HDAC6 can also interact with p53 and attenuate its transcriptional activity through the promotion of its degradation [37][20]. p53 is mutated in 43% of CRC cases [97][71], indicating that the induction of the degradation of mutant p53 may represent a potential therapeutic approach. In this sense, several Hsp90 and HDAC inhibitors have been shown to destabilize p53 mutant proteins. Hsp90 inactivates the p53 E3 ubiquitin ligases MDM2 and CHIP, thereby increasing mutant p53 levels, while Hsp90 chaperone activity is enhanced by HDAC6-mediated deacetylation [98,99][72][73]. Thus, targeting of Hsp90 or HDAC6 induces the degradation of mutant p53. Whereas several Hsp90 inhibitors have so far only been investigated in clinical trials, HDAC inhibitors have already been approved by the Food and Drug Administration (FDA) for use in cancer therapy [37,97,100][20][71][74]. Due to the increased demand for the degradation of proteins in cancer, proteasomes play an important role in the maintenance of homeostasis. It has been reported that during the progression of CRC, the level of ubiquitin-conjugating enzymes (E2) that are involved in various tumor-promoting processes, specifically the newly identified UBE2Q1, is elevated. UBE2Q1 suppresses the transcriptional activities of p53 by inducing its ubiquitination and degradation and may thereby contribute to the survival of tumor cells [101][75]. Due to frequent p53 mutations in CRC, the apoptotic effect of HDAC inhibition is not always p53 dependent [102][76]. Recent studies indicate that the deacetylation of the DNA repair protein Ku70 by HDAC6 induces its binding to proapoptotic proteins Bax and Mcl-1 in the cytoplasm, increasing their stability and protecting cells from apoptosis [103][77]. However, in vitro treatments of CRC cell lines with HDAC6 inhibitors resulted in increased acetylation of Ku70 and induction of apoptosis by releasing Bax, which was subsequently translocated to mitochondria and induced cytochrome release [25,104][8][78]. Interestingly, the dissociation of the complexes formed between Ku70 and antiapoptotic FLIP protein following the acetylation of Ku70 was found to trigger FLIP polyubiquitination and degradation in proteasomes in CRC cells [39][22]. Furthermore, HDAC6 regulates apoptosis by the deacetylation of the antiapoptotic protein survivin, promoting its exit from the nucleus, which inhibits apoptosis of colon cancer cells [40,41][23][24]. Runt-related transcription factor-2 (Runx2), although initially defined as a transcription factor responsible for osteogenic differentiation in mammals, is closely related to proliferation, invasion, and bone metastasis of multiple cancer types. Interaction between Runx2 and HDAC6 leads to the recruitment of HDAC6 from the cytoplasm to chromatin and repression of the p21 gene promoter that induces proliferation [58][79]. In CRC cells with high Wnt signaling activity, Runx2 was designated as a critical transcription factor to trigger the expression of genes that regulate the epithelial-to-mesenchymal transition in vitro through the orchestration of chromatin organization [105][80]. Moreover, clinical data showed that Runx2 is closely related to an advanced stage of disease and liver metastasis in CRC patients and is associated with shorter survival [106][81].7. Regulation of Signal Transduction Molecules
HDAC6 is also involved in the regulation of the RAS/RAF/MEK/ERK and PI3K/AKT/mTOR signaling pathways. It was first discovered that ERK1 phosphorylates and activates HDAC6 [60][82]. Further investigations revealed that HDAC6 deacetylates ERK1/2 and augments its activity while acetylation by CREB-binding protein and p300 decreases its activity toward the transcription factor ELK1. Accordingly, HDAC6 inhibition has been shown to suppress tumor proliferation and induce apoptosis via the deactivation of AKT and ERK signaling [48][31]. Moreover, HDAC6 affects AKT signaling indirectly by deacetylating Hsp90, which subsequently binds to AKT, protecting it from phosphatases and preserving its activity. By stimulating the AKT signaling pathway, HDAC6 contributes to cancer cell migration and angiogenesis [107][83]. HDAC6 inhibition has been shown to decrease AKT binding to PIP3 and activity [49][32]. This is relevant for CRC as this tumor may show increased activation of this signaling pathway as well as increased expression of HDAC6 [108][84]. HDAC6 regulates EGFR endocytosis and degradation by controlling the acetylation status of α-tubulin and subsequently receptor trafficking along microtubules. A negative feedback loop consisting of EGFR-mediated phosphorylation of HDAC6 on Tyr570 reduces the deacetylase activity and increases the acetylation of α-tubulin [109][85].8. Regulation of NLRP3 Inflammasome
HDAC6 is involved in the assembly, priming, and activation of inflammasomes, the cytoplasmic protein complexes that are a crucial part of the innate immune system. The NLRP3 inflammasome contains an NLRP3 pattern recognition receptor (PRR), an apoptosis-associated spike-like protein containing a caspase recruitment domain for caspase-1 (ASC), and caspase-1 itself. In response to the activation of PRR by the products of damaged or dying cancer cells and activation of the NF-κB transcription factor, inflammasomes induce expression of NLRP3, pro-IL-1β, and pro-IL-18 [110][86]. This is followed by caspase-1-induced maturation of the proactive inflammatory cytokines to IL-1β and IL-18 and subsequent cleaving of gasdermin D to induce pyroptosis [80][51]. HDAC6 facilitates the priming of the NLRP3 inflammasome most prominently by deacetylating and activating the p65 subunit of NF-κB that subsequently induces the transcription of genes for NLRP3, pro-IL-1β, and pro-IL-18 [111][87]. Moreover, HDAC6 induces the activation of inflammasomes by activating PrxII, which increases the level of reactive oxygen species, which are important activators of inflammasomes [112][88]. It has been shown that HDAC6 inhibition upregulates p65 expression in the cytoplasm and reduces p65 expression in the nuclei of macrophages to attenuate the transcription of NLRP3 and reduce pyroptosis [113][89]. However, HDAC6 negatively regulates inflammasome activation through its interaction with ubiquitinated NLRP3 [80][51]. NLRP3 can impact CRC development due to its broad activity in shaping immune responses, apoptosis, and the gut microbiome. In this sense, the role of inflammasomes in colitis and colitis-associated CRC has been shown in animal models [114,115][90][91]. Moreover, the activation of the NLRP3 inflammasome in macrophages has been shown to promote the invasion of CRC cells by regulating the epithelial–mesenchymal transition via secretion of IL-1β from activated macrophages [115,116][91][92]. This finding, together with a clinical finding that suggested the positive correlation of NLRP3 expression with advanced disease and poor prognosis in patients with CRC, indicates the potential relevance of NLRP3 inflammasome as a therapeutic target [117][93].9. Role of HDAC6 in Tumor Invasiveness
HDAC6 is involved in multiple phases of the epithelial–mesenchymal transition (EMT) by which tumor cells lose epithelial characteristics, cell-to-cell junctions, reorganize their cytoskeleton, increase cell motility, and acquire properties that are typical of mesenchymal cells. HDAC6 plays an important role in the EMT by deacetylating α-tubulin and augmenting cell motility. Moreover, a high level of acetylated α-tubulin was correlated with epithelial morphology, while the deacetylated form corresponded to EMT transition [118][94]. The most potent inducer of EMT, transforming growth factor (TGF)-β, has the ability to activate HDAC6. TGF-β and HDAC6 pathways intercept and induce phosphorylation and activation of the Smad3 molecule that inhibits the transcription of E-cadherin, leading to a mesenchymal-like phenotype of malignant cells [43][26]. It has been suggested that HDAC6 may induce the activity of the signal transducer Smad3 molecule directly by deacetylation [44][27], or indirectly by deacetylating α-tubulin and promoting Smad3 release [119][95]. Moreover, selective HDAC6 inhibition was reported to downregulate the expression of TGF-βRⅠ and the phosphorylation of Smad3 and EMT-inducing transcription factor Snail that led to the preserved expression of E-cadherin in cultured cancer cells [118,120][94][96]. IL-6 has been reported to induce the expression of HDAC6, concomitantly with increased proliferation, migration, and EMT of tumor cells. Moreover, the IL-6-induced HDAC6 not only upregulated the IL-6 downstream JAK2/STAT3 pathway but also co-activated TGF-β/Smad3 signaling [121][97]. The increased level of circulating IL-6 has been related to metastatic disease and poor prognostic outcome in cancer patients with diverse histological tumor types, as reported in numerous studies [122,123,124][98][99][100]. Through deacetylation of cortactin and tubulin, HDAC6 is involved in the formation of invadopodia, the actin-rich proteolytic structures specialized in the degradation of the extracellular matrix that mediates the invasion of malignant cells to distant tissues and organs [125][101]. Invadopodia have been observed during metastatic invasion of CRC [126][102]. The hypoxic conditions that prevail in solid tumors enhance HDAC6 deacetylase activity by EGFR, resulting in enhanced Smad phosphorylation and nuclear accumulation that influence invadopodia formation [127,128][103][104]. Therefore, considering the role of HDAC6 in hypoxia-induced metastatic invasion to regional lymphatics, the therapeutic targeting of HDAC6 may have important therapeutic implications for the treatment of metastatic disease [128,129][104][105]. Due to its role in the regulation of cytoskeletal dynamics, HDAC6 contributes to angiogenesis by regulating the polarization and migration of vascular endothelial cells in a microtubule end-binding protein (EB) 1-dependent manner and generating capillary-like structures [130][106]. In support of this, the upregulation of HDAC6 mRNA levels and protein levels has been shown in endothelial cells under hypoxic conditions [131][107]. Furthermore, HDAC6 was found to associate with HIF-1α [132][108] and with its transcriptional target, the VEGF receptor, thereby increasing their stability and activity in cancer cells [133][109]. Moreover, it was reported that HDAC6 affects the Hsp90-mediated regulation of VEGFR in tumor cells [134][110]. HDAC6 modifies β-catenin, which plays an essential role in cell-to-cell adherens junctions as it links E-cadherin to actin filaments. β-catenin is a key player in the Wnt cascade signaling pathway that induces EMT, cancer cell motility, and cancer stem cell maintenance [135][111]. The activation of HDAC6 upon simulation of EGFR leads to the deacetylation of β-catenin as well as the breakup of cell-to-cell junctions, which increases the level of nuclear β-catenin, either by direct release of the junctional β-catenin from the cell membrane or by activating E-cadherin endocytosis. Nuclear localization of β-catenin increases the proliferative potential of tumor cells by activation of target genes for c-myc and cyclin D1 [45][28]. It was reported that HDAC6 negatively regulates EGFR endocytosis and degradation in lysosomes by controlling the acetylation status of α-tubulin and hence the receptor trafficking along microtubules. However, the phosphorylation of HDAC6 by activated EGFR was found to reduce deacetylase activity and create a negative feedback loop, leading to increased degradation of activated EGFR [109][85]. Wnt/β-catenin signaling is involved in the tumorigenesis of CRC. The presence of mutations in APC (adenomatous polyposis coli) induces the nuclear localization of β-catenin and expression of Wnt target genes that promote tumor progression in CRC [45,136][28][112].10. Involvement of HDAC6 in Immune Responses
HDAC6 has been shown to intervene in many aspects of the innate and adaptive immune responses. It affects antigen (Ag) uptake and presentation by antigen-presenting cells (APCs), dendritic cells (DCs), or macrophages and the cytotoxic function of natural killer (NK) cells, thus influencing the innate immune cells. Furthermore, HDAC6 partially affects T-cell activation and antitumor cytotoxicity in adaptive immune responses [137][113]. HDAC6 displays many of its immune-related effects by affecting the signal transducer and activator of transcription (STAT)3 signaling pathway, which is involved in the development of malignancies and in the induction and maintenance of immune tolerance and inhibition of immune responses [46,137][29][113]. STAT3 signaling can be activated with cytokines interleukin (IL)-6, IL-10, IL-21, and tumor necrosis factor (TNF) that in DCs downregulate the expression of MHC class II and costimulatory molecules, thereby inducing the tolerogenic immune response [138][114]. Furthermore, STAT3 plays a key role in suppressing signal transduction mediated by Toll-like receptors (TLRs) in mature phagocytic cells. Accordingly, it has been shown that Stat3-deficient macrophages and DCs produce increased levels of proinflammatory cytokines (TNF, IL-1β, IL-6, IL-12) which activate the immune response upon TLR4 activation while reducing the amount of anti-inflammatory IL-10 and losing responsiveness to this cytokine that inhibits TLR4-dependent pro-inflammatory cytokine production [139][115]. The relevance of HDAC6–STAT3 signaling in immunomodulatory pathways in CRC has been confirmed by the pharmacological inhibition of HDAC6 that led to reduced functionality of STAT3 signaling, impacting the expression of genes involved in the inflammatory and immune responses [140][116]. HDAC6 is also involved in the regulation of macrophages [141][117]. In this sense, after stimulation of macrophages with lipopolysaccharide (LPS), HDAC6 was shown to translocate to the cell periphery where it induced cortactin deacetylation and the formation of invadopodia protrusions that increase cell mobility and enable infiltration into tissues [142][118]. Under physiological conditions, macrophages activated by LPS, IFN-γ, or TNF polarize into the M1 type that secretes pro-inflammatory cytokines (TNF, IL-1β, IL-6, and IL-12) and activate the antitumor immune response. Conversely, in tumors, after exposure to immunosuppressive cytokines IL-10 and TGF-β in the tumor microenvironment (TME), macrophages often differentiate into the M2 type, which is known to produce high concentrations of the immunosuppressive cytokine IL-10 [137][113]. However, HDAC6 inhibition in macrophages and DCs results in diminished production of IL-10 and enables them to effectively activate Ag-specific naive T cells. HDAC6 forms a molecular complex with STAT3 that has been detected in cytoplasmic and nuclear compartments of APCs [46][29]. It has been reported that colon cancer specimens with high HDAC6 expression show increased infiltration of immunosuppressive M2 macrophages that can be attributed to HDAC6 activity [47][30]. In this setting, HDAC6 deacetylates TGF-β-activated kinase 1 (TAK1), which subsequently activates p38 MAPK, leading to phosphorylation and activation of A disintegrin and metalloproteinase-17 (ADAM17). ADAM17, through its proteolytic activity, is responsible for the shedding of IL-6 receptor [47][30] from the cell membrane, resulting in the release of soluble IL-6 receptor (sIL-6R). Aside from the classical IL-6 signaling that involves IL-6 ligation to membrane-bound IL-6R and gp130 transmembrane receptor dimerization, “IL-6 trans-signaling” is mediated by sIL-6R, which forms a complex with IL-6 and directly engages gp130 [121][97]. Since ADAM17 is more abundant in CRC cells compared to normal tissue, it contributes to increased levels of soluble IL-6R that promote M2 macrophage polarization [47,143][30][119]. As HDAC6 is involved in the intracellular trafficking of granules, HDAC6-deficient CD8+ cytotoxic T lymphocytes (CTLs) were reported to display defective in vitro cytolytic activity due to altered dynamics, inhibited transport of lytic granules to the immune synapse, and deficient exocytosis, while target cell recognition, T cell receptor (TCR) activation, and IFN-γ production were not inhibited [144][120]. HDAC6 affects the development and activity of regulatory T cells (Treg) that have the physiological function of suppressing excessive immune responses to maintain immune homeostasis. In tumor immunity, Tregs are involved in tumor development and progression as they impair T cell function through the secretion of immune suppressive cytokines (IL-10, TGF-β, IL-35), consumption of IL-2 that leads to its depletion in the TME, and expression of inhibitory checkpoint receptor cytotoxic T lymphocyte-associated protein (CTLA-4). HDAC6 inhibits Treg differentiation as it deacetylates the Foxp3 transcription factor and inhibits the transcription of Foxp3-induced genes. It has been reported in several murine models that treatment with HDAC6-specific inhibitors increased the activity and induced the differentiation of Tregs [145,146][121][122]. Inhibition of HDAC6 by tubastatin A increased the acetylation of Hsp90 in Tregs, inducing the release of HSF-1 and upregulation of Treg-related genes [146][122]. Given that acetylation reduces the proteasomal degradation of FoxP3, HDAC6 inhibition would be expected to increase FoxP3 expression and increase the Treg number or function [147,148][123][124]. Regardless of these findings, there are conflicting reports on the role of HDAC6 in Treg cells. In this sense, one study reported that pharmacological inhibition of HDAC6 inhibited Treg cell differentiation and suppressive function in TGF-β-induced murine Treg cell differentiation by inhibiting their proliferation [149][125]. It has not yet been elucidated why different studies reported different effects of HDAC6 inhibition on Tregs. Increased accumulation of Treg cells is generally associated with CRC progression and metastasis, immunotherapy failure, and a poorer prognosis, although this correlation is not conclusive [150][126]. HDAC6 activity in tumor cells and immune cells in the TME has been shown to regulate the expression of tumor-associated antigens, MHC class I molecules, costimulatory molecules, and cytokines [151][127].References
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873.
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase 6 in health and disease. Epigenomics 2015, 7, 103–118.
- Hai, Y.; Christianson, D.W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747.
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013, 280, 775–793.
- García-Domínguez, D.J.; Hontecillas-Prieto, L.; Kaliszczak, M.; He, M.; Burguillos, M.A.; Bekay, R.; Abdul-Salam, V.B.; Khozoie, C.; Shah, K.; O’Neill, K.; et al. Novel nuclear role of HDAC6 in prognosis and therapeutic target for colorectal cancer. bioRxiv 2020.
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179.
- Pulya, S.; Amin, S.A.; Adhikari, N.; Biswas, S.; Jha, T.; Ghosh, B. HDAC6 as privileged target in drug discovery: A perspective. Pharmacol. Res. 2021, 163, 105274.
- Subramanian, C.; Jarzembowski, J.A.; Opipari, A.W., Jr.; Castle, V.P.; Kwok, R.P. HDAC6 deacetylates Ku70 and regulates Ku70-Bax binding in neuroblastoma. Neoplasia 2011, 13, 726–734.
- Liu, Y.; Peng, L.; Seto, E.; Huang, S.; Qiu, Y. Modulation of histone deacetylase 6 (HDAC6) nuclear import and tubulin deacetylase activity through acetylation. J. Biol. Chem. 2012, 287, 29168–29174.
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458.
- Zhang, Y.; Kwon, S.; Yamaguchi, T.; Cubizolles, F.; Rousseaux, S.; Kneissel, M.; Cao, C.; Li, N.; Cheng, H.L.; Chua, K.; et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell. Biol. 2008, 28, 1688–1701.
- Valenzuela-Fernández, A.; Cabrero, J.R.; Serrador, J.M.; Sánchez-Madrid, F. HDAC6: A key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008, 18, 291–297.
- Zhang, X.; Yuan, Z.; Zhang, Y.; Yong, S.; Salas-Burgos, A.; Koomen, J.; Olashaw, N.; Parsons, J.T.; Yang, X.J.; Dent, S.R.; et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol. Cell. 2007, 27, 197–213.
- Zhang, J.; Wang, J.; Zhou, Z.; Park, J.E.; Wang, L.; Wu, S.; Sun, X.; Lu, L.; Wang, T.; Lin, Q.; et al. Importance of TFEB acetylation in control of its transcriptional activity and lysosomal function in response to histone deacetylase inhibitors. Autophagy 2018, 14, 1043–1059.
- Zhang, J.; Ng, S.; Wang, J.; Zhou, J.; Tan, S.H.; Yang, N.; Lin, Q.; Xia, D.; Shen, H.M. Histone Deacetylase Inhibitors Induce Autophagy Through FOXO1-Dependent Pathways. Autophagy 2015, 11, 629–642.
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell. 2005, 18, 601–607.
- Li, Z.; Zhuang, M.; Zhang, L.; Zheng, X.; Yang, P.; Li, Z. Acetylation modification regulates GRP78 secretion in colon cancer cells. Sci. Rep. 2016, 7, 30406.
- Yang, C.J.; Liu, Y.P.; Dai, H.Y.; Shiue, Y.L.; Tsai, C.J.; Huang, M.S.; Yeh, Y.T. Nuclear HDAC6 inhibits invasion by suppressing NF-κB/MMP2 and is inversely correlated with metastasis of non-small cell lung cancer. Oncotarget 2015, 6, 30263–30276.
- Ryu, H.W.; Shin, D.H.; Lee, D.H.; Choi, J.; Han, G.; Lee, K.Y.; Kwon, S.H. HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett. 2017, 391, 162–171.
- Ding, G.; Liu, H.D.; Huang, Q.; Liang, H.X.; Ding, Z.H.; Liao, Z.J.; Huang, G. HDAC6 promotes hepatocellular carcinoma progression by inhibiting P53 transcriptional activity. FEBS Lett. 2013, 587, 880–886.
- Mrakovcic, M.; Bohner, L.; Hanisch, M.; Fröhlich, L.F. Epigenetic Targeting of Autophagy via HDAC Inhibition in Tumor Cells: Role of p53. Int. J. Mol. Sci. 2018, 19, 3952.
- Kerr, E.; Holohan, C.; McLaughlin, K.M.; Majkut, J.; Dolan, S.; Redmond, K.; Riley, J.; McLaughlin, K.; Stasik, I.; Crudden, M.; et al. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death Differ. 2012, 19, 1317–1327.
- Riolo, M.T.; Cooper, Z.A.; Holloway, M.P.; Cheng, Y.; Bianchi, C.; Yakirevich, E.; Ma, L.; Chin, Y.E.; Altura, R.A. Histone deacetylase 6 (HDAC6) deacetylates survivin for its nuclear export in breast cancer. J. Biol. Chem. 2012, 287, 10885–10893.
- Jin, J.S.; Tsao, T.Y.; Sun, P.C.; Yu, C.P.; Tzao, C. SAHA inhibits the growth of colon tumors by decreasing histone deacetylase and the expression of cyclin D1 and survivin. Pathol. Oncol. Res. 2012, 18, 713–720.
- Parmigiani, R.B.; Xu, W.S.; Venta-Perez, G.; Erdjument-Bromage, H.; Yaneva, M.; Tempst, P.; Marks, P.A. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 9633–9638.
- Shan, B.; Yao, T.P.; Nguyen, H.T.; Zhuo, Y.; Levy, D.R.; Klingsberg, R.C.; Tao, H.; Palmer, M.L.; Holder, K.N.; Lasky, J.A. Requirement of HDAC6 for transforming growth factor-beta1-induced epithelial-mesenchymal transition. J. Biol. Chem. 2008, 283, 21065–21073.
- Osseni, A.; Ravel-Chapuis, A.; Belotti, E.; Scionti, I.; Gangloff, Y.G.; Moncollin, V.; Mazelin, L.; Mounier, R.; Leblanc, P.; Jasmin, B.J.; et al. Pharmacological inhibition of HDAC6 improves muscle phenotypes in dystrophin-deficient mice by downregulating TGF-β via Smad3 acetylation. Nat. Commun. 2022, 13, 7108.
- Li, Y.; Zhang, X.; Polakiewicz, R.D.; Yao, T.P.; Comb, M.J. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J. Biol. Chem. 2008, 283, 12686–12690.
- Cheng, F.; Lienlaf, M.; Wang, H.W.; Perez-Villarroel, P.; Lee, C.; Woan, K.; Rock-Klotz, J.; Sahakian, E.; Woods, D.; Pinilla-Ibarz, J.; et al. A novel role for histone deacetylase 6 in the regulation of the tolerogenic STAT3/IL-10 pathway in APCs. J. Immunol. 2014, 193, 2850–2862.
- Xu, G.; Niu, L.; Wang, Y.; Yang, G.; Zhu, X.; Yao, Y.; Zhao, G.; Wang, S.; Li, H. HDAC6-dependent deacetylation of TAK1 enhances sIL-6R release to promote macrophage M2 polarization in colon cancer. Cell Death Dis. 2022, 13, 888.
- Wu, J.Y.; Xiang, S.; Zhang, M.; Fang, B.; Huang, H.; Kwon, O.K.; Zhao, Y.; Yang, Z.; Bai, W.; Bepler, G.; et al. Histone deacetylase 6 (HDAC6) deacetylates extracellular signal-regulated kinase 1 (ERK1) and thereby stimulates ERK1 activity. J. Biol. Chem. 2018, 293, 1976–1993.
- Iaconelli, J.; Lalonde, J.; Watmuff, B.; Liu, B.; Mazitschek, R.; Haggarty, S.J.; Karmacharya, R. Lysine Deacetylation by HDAC6 Regulates the Kinase Activity of AKT in Human Neural Progenitor Cells. ACS Chem. Biol. 2017, 12, 2139–2148.
- Chuma, M.; Sakamoto, M.; Yasuda, J.; Fujii, G.; Nakanishi, K.; Tsuchiya, A.; Ohta, T.; Asaka, M.; Hirohashi, S. Overexpression of cortactin is involved in motility and metastasis of hepatocellular carcinoma. J. Hepatol. 2004, 41, 629–636.
- Zhang, X.; Liu, K.; Zhang, T.; Wang, Z.; Qin, X.; Jing, X.; Wu, H.; Ji, X.; He, Y.; Zhao, R. Cortactin promotes colorectal cancer cell proliferation by activating the EGFR-MAPK pathway. Oncotarget 2017, 8, 1541–1554.
- Boyault, C.; Zhang, Y.; Fritah, S.; Caron, C.; Gilquin, B.; Kwon, S.H.; Garrido, C.; Yao, T.P.; Vourc’h, C.; Matthias, P.; et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007, 21, 2172–2181.
- Seigneurin-Berny, D.; Verdel, A.; Curtet, S.; Lemercier, C.; Garin, J.; Rousseaux, S.; Khochbin, S. Identification of components of the murine histone deacetylase 6 complex: Link between acetylation and ubiquitination signaling pathways. Mol. Cell. Biol. 2001, 21, 8035–8044.
- Johnston, H.E.; Samant, R.S. Alternative systems for misfolded protein clearance: Life beyond the proteasome. FEBS J. 2021, 288, 4464–4487.
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980.
- Watabe, M.; Nakaki, T. Protein kinase CK2 regulates the formation and clearance of aggresomes in response to stress. J. Cell Sci. 2011, 124, 1519–1532.
- Watanabe, Y.; Tanaka, M. p62/SQSTM1 in autophagic clearance of a non-ubiquitylated substrate. J. Cell Sci. 2011, 124, 2692–2701.
- Fusco, C.; Micale, L.; Egorov, M.; Monti, M.; D’Addetta, E.V.; Augello, B.; Cozzolino, F.; Calcagnì, A.; Fontana, A.; Polishchuk, R.S.; et al. The E3-ubiquitin ligase TRIM50 interacts with HDAC6 and p62, and promotes the sequestration and clearance of ubiquitinated proteins into the aggresome. PLoS ONE 2012, 7, e40440.
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29.
- Yan, J.; Seibenhener, M.L.; Calderilla-Barbosa, L.; Diaz-Meco, M.T.; Moscat, J.; Jiang, J.; Wooten, M.W.; Wooten, M.C. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS ONE 2013, 8, e76016.
- Boyault, C.; Gilquin, B.; Zhang, Y.; Rybin, V.; Garman, E.; Meyer-Klaucke, W.; Matthias, P.; Müller, C.W.; Khochbin, S. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 2006, 25, 3357–3366.
- Mohamed, A.; Ayman, A.; Deniece, J.; Wang, T.; Kovach, C.; Siddiqui, M.T.; Cohen, C. P62/Ubiquitin IHC expression correlated with clinicopathologic parameters and outcome in gastrointestinal carcinomas. Front. Oncol. 2015, 5, 70.
- Schmitz, K.J.; Ademi, C.; Bertram, S.; Schmid, K.W.; Baba, H.A. Prognostic relevance of autophagy-related markers LC3, p62/sequestosome 1, Beclin-1 and ULK1 in colorectal cancer patients with respect to KRAS mutational status. World J. Surg. Oncol. 2016, 14, 189.
- Passaro, E.; Papulino, C.; Chianese, U.; Toraldo, A.; Congi, R.; Del Gaudio, N.; Nicoletti, M.M.; Benedetti, R.; Altucci, L. HDAC6 Inhibition Extinguishes Autophagy in Cancer: Recent Insights. Cancers 2021, 13, 6280.
- Li, C.; Wang, X.; Li, X.; Qiu, K.; Jiao, F.; Liu, Y.; Kong, Q.; Liu, Y.; Wu, Y. Proteasome Inhibition Activates Autophagy-Lysosome Pathway Associated With TFEB Dephosphorylation and Nuclear Translocation. Front. Cell Dev. Biol. 2019, 22, 170.
- Brijmohan, A.S.; Batchu, S.N.; Majumder, S.; Alghamdi, T.A.; Thieme, K.; McGaugh, S.; Liu, Y.; Advani, S.L.; Bowskill, B.B.; Kabir, M.G.; et al. HDAC6 Inhibition Promotes Transcription Factor EB Activation and Is Protective in Experimental Kidney Disease. Front. Pharmacol. 2018, 9, 34.
- Jung, K.H.; Noh, J.H.; Kim, J.K.; Eun, J.W.; Bae, H.J.; Chang, Y.G.; Kim, M.G.; Park, W.S.; Lee, J.Y.; Lee, S.Y.; et al. Histone deacetylase 6 functions as a tumor suppressor by activating c-Jun NH2-terminal kinase-mediated beclin 1-dependent autophagic cell death in liver cancer. Hepatology 2012, 56, 644–657.
- Chang, P.; Li, H.; Hu, H.; Li, Y.; Wang, T. The Role of HDAC6 in Autophagy and NLRP3 Inflammasome. Front. Immunol. 2021, 12, 763831.
- Kerdiles, Y.M.; Stone, E.L.; Beisner, D.R.; McGargill, M.A.; Ch’en, I.L.; Stockmann, C.; Katayama, C.D.; Hedrick, S.M. Foxo transcription factors control regulatory T cell development and function. Immunity 2010, 33, 890–904.
- Huang, R.; Liu, W. Identifying an Essential Role of Nuclear LC3 for Autophagy. Autophagy 2015, 11, 852–853.
- Liu, K.P.; Zhou, D.; Ouyang, D.Y.; Xu, L.H.; Wang, Y.; Wang, L.X.; Pan, H.; He, X.H. LC3B-II deacetylation by histone deacetylase 6 is involved in serum-starvation-induced autophagic degradation. Biochem. Biophys. Res. Commun. 2013, 441, 970–975.
- Xie, R.; Nguyen, S.; McKeehan, W.L.; Liu, L. Acetylated microtubules are required for fusion of autophagosomes with lysosomes. BMC Cell Biol. 2010, 11, 89.
- Goulielmaki, M.; Koustas, E.; Moysidou, E.; Vlassi, M.; Sasazuki, T.; Shirasawa, S.; Zografos, G.; Oikonomou, E.; Pintzas, A. BRAF associated autophagy exploitation: BRAF and autophagy inhibitors synergise to efficiently overcome resistance of BRAF mutant colorectal cancer cells. Oncotarget 2016, 7, 9188–9221.
- Liu, P.; Xiao, J.; Wang, Y.; Song, X.; Huang, L.; Ren, Z.; Kitazato, K.; Wang, Y. Posttranslational modification and beyond: Interplay between histone deacetylase 6 and heat-shock protein 90. Mol. Med. 2021, 27, 110.
- Tan, Y.; Ci, Y.; Dai, X.; Wu, F.; Guo, J.; Liu, D.; North, B.J.; Huo, J.; Zhang, J. Cullin 3SPOP ubiquitin E3 ligase promotes the poly-ubiquitination and degradation of HDAC6. Oncotarget 2017, 8, 47890–47901.
- Li, Z.Y.; Zhang, C.; Zhang, Y.; Chen, L.; Chen, B.D.; Li, Q.Z.; Zhang, X.J.; Li, W.P. A novel HDAC6 inhibitor Tubastatin A: Controls HDAC6-p97/VCP-mediated ubiquitination-autophagy turnover and reverses Temozolomide-induced ER stress-tolerance in GBM cells. Cancer Lett. 2017, 39, 89–99.
- Zhang, S.; Guo, S.; Li, Z.; Li, D.; Zhan, Q. High expression of HSP90 is associated with poor prognosis in patients with colorectal cancer. PeerJ 2019, 31, e7946.
- Moser, C.; Lang, S.A.; Stoeltzing, O. Heat-shock protein 90 (Hsp90) as a molecular target for therapy of gastrointestinal cancer. Anticancer Res. 2009, 29, 2031–2042.
- Sergi, C.M. Targeting the ‘garbage-bin’ to fight cancer: HDAC6 inhibitor WT161 has an anti-tumor effect on osteosarcoma and synergistically interacts with 5-FU. Biosci. Rep. 2021, 41, BSR20210952.
- Mhaidat, N.M.; Alzoubi, K.H.; Khabour, O.F.; Banihani, M.N.; Al-Balas, Q.A.; Swaidan, S. GRP78 regulates sensitivity of human colorectal cancer cells to DNA targeting agents. Cytotechnology 2016, 68, 459–467.
- Li, Z.; Li, Z. Glucose regulated protein 78: A critical link between tumor microenvironment and cancer hallmarks. Biochim. Biophys. Acta 2012, 1826, 13–22.
- Zhang, S.L.; Zhu, H.Y.; Zhou, B.Y.; Chu, Y.; Huo, J.R.; Tan, Y.Y.; Liu, D.L. Histone deacetylase 6 is overexpressed and promotes tumor growth of colon cancer through regulation of the MAPK/ERK signal pathway. Onco Targets Ther. 2019, 12, 2409–2419.
- Cai, H.; Gong, L.; Liu, J.; Zhou, Q.; Zheng, Z. Diosgenin inhibits tumor angiogenesis through regulating GRP78-mediated HIF-1α and VEGF/VEGFR signaling pathways. Pharmazie 2019, 74, 680–684.
- Harris, S.; Levine, A. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908.
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001, 20, 1331–1340.
- Brooks, C.L.; Gu, W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462.
- Zopf, S.; Neureiter, D.; Bouralexis, S.; Abt, T.; Glaser, K.B.; Okamoto, K.; Ganslmayer, M.; Hahn, E.G.; Herold, C.; Ocker, M. Differential response of p53 and p21 on HDAC inhibitor-mediated apoptosis in HCT116 colon cancer cells in vitro and in vivo. Int. J. Oncol. 2007, 31, 1391–1402.
- Liebl, M.C.; Hofmann, T.G. The Role of p53 Signaling in Colorectal Cancer. Cancers 2021, 13, 2125.
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588.
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913.
- Parrales, A.; Iwakuma, T. Targeting oncogenic mutant p53 for cancer therapy. Front. Oncol. 2015, 5, 288.
- Rasouli, M.; Khakshournia, S.; Vakili, O.; Dastghaib, S.; Seghatoleslam, A.; Shafiee, S.M. The crosstalk between ubiquitin-conjugating enzyme E2Q1 and p53 in colorectal cancer: An in vitro analysis. Med. Oncol. 2023, 40, 199.
- Hirose, T.; Sowa, Y.; Takahashi, S.; Saito, S.; Yasuda, C.; Shindo, N.; Furuichi, K.; Sakai, T. p53-independent induction of Gadd45 by histone deacetylase inhibitor: Coordinate regulation by transcription factors Oct-1 and NF-Y. Oncogene 2003, 22, 7762–7773.
- Gong, P.; Wang, Y.; Jing, Y. Apoptosis Induction by Histone Deacetylase Inhibitors in Cancer Cells: Role of Ku70. Int. J. Mol. Sci. 2019, 20, 1601.
- Meng, J.; Zhang, F.; Zhang, X.T.; Zhang, T.; Li, Y.H.; Fan, L.; Sun, Y.; Zhang, H.L.; Mei, Q.B. Ku70 is essential for histone deacetylase inhibitor trichostatin A-induced apoptosis. Mol. Med. Rep. 2015, 2, 581–586.
- Westendorf, J.J.; Zaidi, S.K.; Cascino, J.E.; Kahler, R.; van Wijnen, A.J.; Lian, J.B.; Yoshida, M.; Stein, G.S.; Li, X. Runx2 (Cbfa1, AML-3) interacts with histone deacetylase 6 and represses the p21(CIP1/WAF1) promoter. Mol. Cell. Biol. 2002, 22, 7982–7992.
- Yi, H.; Li, G.; Long, Y.; Liang, W.; Cui, H.; Zhang, B.; Tan, Y.; Li, Y.; Shen, L.; Deng, D.; et al. Integrative multi-omics analysis of a colon cancer cell line with heterogeneous Wnt activity revealed RUNX2 as an epigenetic regulator of EMT. Oncogene 2020, 39, 5152–5164.
- Lin, T.-C. RUNX2 and Cancer. Int. J. Mol. Sci. 2023, 24, 7001.
- Williams, K.A.; Zhang, M.; Xiang, S.; Hu, C.; Wu, J.Y.; Zhang, S.; Ryan, M.; Cox, A.D.; Der, C.J.; Fang, B.; et al. Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J. Biol. Chem. 2013, 288, 33156–33170.
- Tsutsumi, S.; Beebe, K.; Neckers, L. Impact of heat-shock protein 90 on cancer metastasis. Future Oncol. 2009, 5, 679–688.
- Johnson, S.M.; Gulhati, P.; Rampy, B.A.; Han, Y.; Rychahou, P.G.; Doan, H.Q.; Weiss, H.L.; Evers, B.M. Novel expression patterns of PI3K/Akt/mTOR signaling pathway components in colorectal cancer. J. Am. Coll. Surg. 2010, 210, 767–776.
- Deribe, Y.L.; Wild, P.; Chandrashaker, A.; Curak, J.; Schmidt, M.H.H.; Kalaidzidis, Y.; Milutinovic, N.; Kratchmarova, I.; Buerkle, L.; Fetchko, M.J.; et al. Regulation of epidermal growth factor receptor trafficking by lysine deacetylase HDAC6. Sci. Signal. 2009, 2, ra84.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328.
- Moreno-Gonzalo, O.; Ramírez-Huesca, M.; Blas-Rus, N.; Cibrián, D.; Saiz, M.L.; Jorge, I.; Camafeita, E.; Vázquez, J.; Sánchez-Madrid, F. HDAC6 controls innate immune and autophagy responses to TLR-mediated signalling by the intracellular bacteria Listeria monocytogenes. PLoS Pathog. 2017, 13, e1006799.
- Youn, G.S.; Lee, K.W.; Choi, S.Y.; Park, J. Overexpression of HDAC6 Induces Pro-Inflammatory Responses by Regulating ROS-MAPK-NF-Kappab/AP-1 Signaling Pathways in Macrophages. Free Radic. Biol. Med. 2016, 97, 14–23.
- Xu, S.; Chen, H.; Ni, H.; Dai, Q. Targeting HDAC6 attenuates nicotine-induced macrophage pyroptosis via NF-κB/NLRP3 pathway. Atherosclerosis 2021, 317, 1–9.
- Sharma, B.R.; Kanneganti, T.D. Inflammasome signaling in colorectal cancer. Transl. Res. 2023, 252, 45–52.
- Zhang, L.; Wang, Y.; Liu, X.; Zhang, Y. NLRP3 Inflammasome Activation in MΦs-CRC Crosstalk Promotes Colorectal Cancer Metastasis. Ann. Clin. Lab. Sci. 2022, 52, 571–579.
- Vafaei, S.; Taheri, H.; Hajimomeni, Y.; Fakhre Yaseri, A.; Abolhasani Zadeh, F. The role of NLRP3 inflammasome in colorectal cancer: Potential therapeutic target. Clin. Transl. Oncol. 2022, 24, 1881–1889.
- Shi, F.; Wei, B.; Lan, T.; Xiao, Y.; Quan, X.; Chen, J.; Zhao, C.; Gao, J. Low NLRP3 expression predicts a better prognosis of colorectal cancer. Biosci. Rep. 2021, 41, BSR20210280.
- Gu, S.; Liu, Y.; Zhu, B.; Ding, K.; Yao, T.P.; Chen, F.; Zhan, L.; Xu, P.; Ehrlich, M.; Liang, T.; et al. Loss of α-Tubulin Acetylation Is Associated with TGF-β-induced Epithelial-Mesenchymal Transition. J. Biol. Chem. 2016, 291, 5396–5405.
- Dong, C.; Li, Z.; Alvarez, R., Jr.; Feng, X.H. Goldschmidt-Clermont PJ. Microtubule binding to Smads may regulate TGF beta activity. Mol. Cell 2000, 5, 27–34.
- Xu, L.; Liu, N.; Gu, H.; Wang, H.; Shi, Y.; Ma, X.; Ma, S.; Ni, J.; Tao, M.; Qiu, A.; et al. Histone deacetylase 6 inhibition counteracts the epithelial–mesenchymal transition of peritoneal mesothelial cells and prevents peritoneal fibrosis. Oncotarget 2017, 8, 88730–88750.
- Shi, Y.; Tao, M.; Ni, J.; Tang, L.; Liu, F.; Chen, H.; Ma, X.; Hu, Y.; Zhou, X.; Qiu, A.; et al. Requirement of Histone Deacetylase 6 for Interleukin-6 Induced Epithelial-Mesenchymal Transition, Proliferation, and Migration of Peritoneal Mesothelial Cells. Front. Pharmacol. 2021, 12, 722638.
- Mirjačić Martinović, K.; Vuletić, A.; Tišma Miletić, N.; Matković, S.; Gavrilović, D.; Ninković, A.; Jurišić, V.; Babović, N. Circulating IL-6 is associated with disease progression in BRAFwt metastatic melanoma patients receiving anti-PD-1 therapy. J Clin. Pathol. 2023.
- Waldner, M.J.; Foersch, S.; Neurath, M.F. Interleukin-6--a key regulator of colorectal cancer development. Int. J. Biol. Sci. 2012, 8, 1248–1253.
- Vuletić, A.; Mirjačić Martinović, K.; Tišma Miletić, N.; Zoidakis, J.; Castellvi-Bel, S.; Čavić, M. Cross-Talk Between Tumor Cells Undergoing Epithelial to Mesenchymal Transition and Natural Killer Cells in Tumor Microenvironment in Colorectal Cancer. Front. Cell Dev. Biol. 2021, 9, 750022.
- Gould, C.M.; Courtneidge, S.A. Regulation of invadopodia by the tumor microenvironment. Cell Adh. Migr. 2014, 8, 226–235.
- Schoumacher, M.; Goldman, R.D.; Louvard, D.; Vignjevic, D.M. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J. Cell Biol. 2010, 189, 541–556.
- Wang, J.; Lin, A.; Lu, L. Effect of EGF-induced HDAC6 activation on corneal epithelial wound healing. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2943–2948.
- Arsenault, D.; Brochu-Gaudreau, K.; Charbonneau, M.; Dubois, C.M. HDAC6 deacetylase activity is required for hypoxia-induced invadopodia formation and cell invasion. PLoS ONE 2013, 8, e55529.
- Turtoi, A.; Peixoto, P.; Castronovo, V.; Bellahcène, A. Histone deacetylases and cancer-associated angiogenesis: Current understanding of the biology and clinical perspectives. Crit. Rev. Oncog. 2015, 20, 119–137.
- Li, D.; Xie, S.; Ren, Y.; Huo, L.; Gao, J.; Cui, D.; Liu, M.; Zhou, J. Microtubule-associated deacetylase HDAC6 promotes angiogenesis by regulating cell migration in an EB1-dependent manner. Protein Cell 2011, 2, 150–160.
- Kaluza, D.; Kroll, J.; Gesierich, S.; Yao, T.P.; Boon, R.A.; Hergenreider, E.; Tjwa, M.; Rössig, L.; Seto, E.; Augustin, H.G.; et al. Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J. 2011, 30, 4142–4156.
- Qian, D.Z.; Kachhap, S.K.; Collis, S.J.; Verheul, H.M.; Carducci, M.A.; Atadja, P.; Pili, R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006, 66, 8814–8821.
- Ellis, L.; Hammers, H.; Pili, R. Targeting tumor angiogenesis with histone deacetylase inhibitors. Cancer Lett. 2009, 280, 145–153.
- Park, J.H.; Kim, S.H.; Choi, M.C.; Lee, J.; Oh, D.Y.; Im, S.A.; Bang, Y.J.; Kim, T.Y. Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem. Biophys. Res. Commun. 2008, 368, 318–322.
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of β-catenin. EMBO J. 2012, 31, 2714–2736.
- Noe, O.; Filipiak, L.; Royfman, R.; Campbell, A.; Lin, L.; Hamouda, D.; Stanbery, L.; Nemunaitis, J. Adenomatous polyposis coli in cancer and therapeutic implications. Oncol. Rev. 2021, 15, 534.
- McCaw, T.R.; Randall, T.D.; Forero, A.; Buchsbaum, D.J. Modulation of antitumor immunity with histone deacetylase inhibitors. Immunotherapy 2017, 9, 1359–1372.
- Sulczewski, F.B.; Martino, L.A.; Salles, D.; Yamamoto, M.M.; Rosa, D.S.; Boscardin, S.B. STAT3 signaling modulates the immune response induced after antigen targeting to conventional type 1 dendritic cells through the DEC205 receptor. Front. Immunol. 2022, 13, 1006996.
- Hillmer, E.J.; Zhang, H.; Li, H.S.; Watowich, S.S. STAT3 signaling in immunity. Cytokine Growth Factor Rev. 2016, 31, 1–15.
- Mardones, C.; Navarrete-Munoz, C.; Armijo, M.E.; Salgado, K.; Rivas-Valdes, F.; Gonzalez-Pecchi, V.; Farkas, C.; Villagra, A.; Hepp, M.I. Role of HDAC6-STAT3 in immunomodulatory pathways in Colorectal cancer cells. Mol. Immunol. 2023, 164, 98–111.
- Knox, T.; Sahakian, E.; Banik, D.; Hadley, M.; Palmer, E.; Noonepalle, S.; Kim, J.; Powers, J.; Gracia-Hernandez, M.; Oliveira, V.; et al. Selective HDAC6 inhibitors improve anti-PD-1 immune checkpoint blockade therapy by decreasing the anti-inflammatory phenotype of macrophages and down-regulation of immunosuppressive proteins in tumor cells. Sci. Rep. 2019, 9, 6136, Erratum in Sci. Rep. 2019, 9, 14824.
- Zhang, Q.Q.; Zhang, W.J.; Chang, S. HDAC6 inhibition: A significant potential regulator and therapeutic option to translate into clinical practice in renal transplantation. Front. Immunol. 2023, 14, 1168848.
- Chen, L.; Wang, S.; Wang, Y.; Zhang, W.; Ma, K.; Hu, C.; Zhu, H.; Liang, S.; Liu, M.; Xu, N. IL-6 influences the polarization of macrophages and the formation and growth of colorectal tumor. Oncotarget 2018, 9, 17443–17454.
- Nunez-Andrade, N.; Iborra, S.; Trullo, A.; Moreno-Gonzalo, O.; Calvo, E.; Catalán, E.; Menasche, G.; Sancho, D.; Vázquez, J.; Yao, T.P.; et al. HDAC6 regulates the dynamics of lytic granules in cytotoxic T lymphocytes. J. Cell Sci. 2016, 129, 1305–1311.
- Beier, U.H.; Wang, L.; Han, R.; Akimova, T.; Liu, Y.; Hancock, W.W. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci. Signal. 2012, 5, ra45.
- de Zoeten, E.F.; Wang, L.; Butler, K.; Beier, U.H.; Akimova, T.; Sai, H.; Bradner, J.E.; Mazitschek, R.; Kozikowski, A.P.; Matthias, P.; et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol. Cell. Biol. 2011, 31, 2066–2078.
- Xiao, Y.; Li, B.; Zhou, Z.; Hancock, W.W.; Zhang, H.; Greene, M.I. Histone acetyltransferase mediated regulation of FOXP3 acetylation and Treg function. Curr. Opin. Immunol. 2010, 22, 583–591.
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564.
- Lee, J.H.; Kim, H.S.; Jang, S.W.; Lee, G.R. Histone deacetylase 6 plays an important role in TGF-β-induced murine Treg cell differentiation by regulating cell proliferation. Sci. Rep. 2022, 12, 22550.
- Aristin Revilla, S.; Kranenburg, O.; Coffer, P.J. Colorectal Cancer-Infiltrating Regulatory T Cells: Functional Heterogeneity, Metabolic Adaptation, and Therapeutic Targeting. Front. Immunol. 2022, 13, 903564.
- Woan, K.V.; Lienlaf, M.; Perez-Villaroel, P.; Lee, C.; Cheng, F.; Knox, T.; Woods, D.M.; Barrios, K.; Powers, J.; Sahakian, E.; et al. Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: Enhanced antitumor immunity and impaired cell proliferation. Mol. Oncol. 2015, 9, 1447–1457.
More