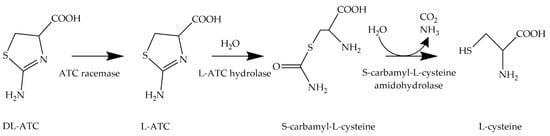
Transformation of DL-ATC to L-cysteine. In this enzymatic three-step process, DL-ATC is isomerized to L-ATC by ATC racemase. Then, L-ATC is hydrolyzed to S-carbamyl-L-cysteine (SCC) by L-ATC hydrolase. Finally, SCC is cleaved into L-cysteine, CO
, and ammonia by S-cabamyl-L-cysteine amidohydrolase.
2. Metabolic Engineering Strategies
Microorganisms use two main pathways for the biosynthesis of L-cysteine. In enteric bacteria such as
E. coli, the precursor of L-cysteine is L-serine
[20]. L-cysteine is synthesized from L-serine in two steps, which involve the substitution of the β-hydroxyl group with a thiol group (
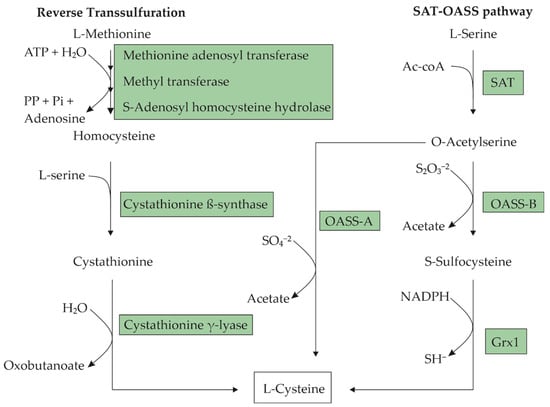
Figure 1). The first step involves the acetylation of the β-hydroxyl of L-serine, resulting in an O-acetyl-L-serine (OAS) catalyzed by L-serine O-acetyltransferase (SAT), which is encoded by the
cysE gene. The second step involves the α,β-elimination of acetate from OAS and the addition of H
2S to give an L-cysteine catalyzed by O-acetyl-L-serine sulfhydrylase-A (OASS-A), which is encoded by the
cysK gene.
Corynebacterium glutamicum and
Pantoea ananantis synthesize L-cysteine through the same pathway as
E. coli [12]. Other microorganisms, such as
Pseudomonas putida [21],
Saccharomyces cerevisiae [22], and
Lactococcus lactis [23] synthesize L-cysteine from L-methionine through the reverse transsulfuration pathway that consists in the cleavage of a methyl group from L-methionine to generate a homocysteine catalyzed by a multienzyme complex, and the homocysteine’s subsequent transformation to cystathionine by the enzyme cystathionine beta-synthase (CBS). L-cysteine can be produced from cystathionine through the enzyme cystathionine gamma-lyase (CSE) (
Figure 1).
The choice of the starting strain for metabolic engineering has an impact on the potential enhancements achieved through the modification of metabolic steps
[24]. This will be shown in an example with
E. coli: It has been established that the transcription levels of key metabolic genes are the major modulators of different glucose utilization pathways in
E. coli [25]. Also, the genetic difference in the catabolite repression protein (CRP) among the
E. coli K-12 strains affects sulfur metabolism
[26]. During the last 20 years, four
E. coli K-12 strains (BW25113, JM109, W3110, and MG1655, respectively) have been utilized in L-cysteine production
[27]. Therefore, the selection of a suitable chassis for L-cysteine production has been deemed the foundation for the successful implementation of metabolic engineering strategies
[28]. To reach a more suitable chassis cell, the L-cysteine-producing plasmid pLH03, containing P
-cysE (coding for serine acetyltransferase) and P
-ydeD (a cysteine exporter), was transformed into the four aforementioned
E. coli K-12 hosts. The results of shake flask experiments (minimal medium with thiosulfate as the sulfur source) showed that the best chassis for the plasmid pLH03 was BW25113, since it produced 345.8 mg L
−1 of L-cysteine, followed by JM109 (190.3 mg L
−1). By contrast, neither W3110 nor MG1655 were deemed suitable base strains for this plasmid, since they each produced less than 70 mg L
−1 product.
[28].
2.1. Biosynthesis of L-Cysteine
The acetylation of L-serine catalyzed by serine acetyltransferase (SAT) is the rate-limiting step of L-cysteine biosynthesis in many microorganisms. The activity of SAT is suppressed by L-cysteine via feedback inhibition (
Figure 2). An approach to enhance the fermentative production of L-cysteine utilizes feedback inhibition-insensitive SATs. These SATs are obtained by the following means:
-
Engineering SAT from
E. coli through site-directed mutagenesis or random mutagenesis
[29][30][29,30];
-
Utilizing natural SAT from higher plants insensitive to feedback inhibition
[31][32][31,32].
Nakamori et al.
[33] obtained feedback inhibition-insensitive SATs by engineering SAT from
E. coli through site-directed mutagenesis using PCR. In this study, the
cysE gene coding for SAT was altered by replacing the methionine at position 256 with other amino acids. The transformed microorganisms, having the altered
cysE gene, produced 50–300 mg L
−1 of L-cysteine plus L-cystine after 72 h of cultivation in shake flasks containing a minimal medium at 30 °C. Furthermore, the transformation of the altered
cysE gene in a mutant strain of
E. coli, JM39-8, with decreased cysteine degradation abilities (10% CD activity) resulted in an increased production to a maximum of 790 mg L
−1 of L-cysteine plus L-cystine in shake flasks under the same conditions as above.
Another approach to obtain feedback inhibition-insensitive SATs could be to use natural SATs from higher plants. In
Arabidopsis thaliana, three cDNA clones encoding SAT isoforms (SAT-c, SAT-p, and SAT-m) have been isolated. Noji et al.
[33] analyzed these SATs and reported that SAT-p and SAT-m were feedback inhibition-insensitive isozymes. However, the activity of SAT-c was feedback inhibited by a low concentration of L-cysteine.
C. glutamicum is a nonpathogenic and high G+C gram-positive bacterium industrially used for decades for the fermentative production of amino acids such as L-glutamic acid and L-lysine
[34][35][35,36]. Joo et al.
[36][37] investigated the effect of the overexpression of genes encoding SAT, OASS, and a transcriptional regulator in
C. glutamicum. The results showed that L-cysteine production was enhanced by approximately three-fold, with the recombinant strain reaching a maximal L-cysteine concentration of approximately 60 mg L
−1 in shake flasks at 30 °C after 15 h of cultivation.
One of the key enzymes in the L-serine biosynthesis pathway, 3-phosphoglycerate dehydrogenase (PGDH), coded by the gene
serA, is feedback inhibited by L-serine
[7][13][7,13]. The authors of
[37][39] converted the 344th and 346th amino acids of
E. coli’s PGDH to alanine, which created a feedback-insensitive
serA.
2.2. Weakening the Degradation of L-Cysteine
It is well known that disrupting the degradation pathway and enhancing metabolic fluxes for the desired amino acids are effective approaches to improve their production
[19][37][38][39][19,39,40,41]. The degradation of L-cysteine to pyruvate, ammonia, and sulfide is mainly catalyzed by L-cysteine desulfhydrase (CD). Wada et al.
[40][42] purified and characterized CD from
C. glutamicum. After comparing the partial amino acid sequence, it was identified that the enzyme CD is a C-S lyase with α,β-elimination activity, and it is encoded by the
aecD gene
[41][43]. They showed that the
aecD gene product is involved in L-cysteine degradation, and the disruption of the
aecD gene resulted in an increased production of L-cysteine (approximately 290 mg L
−1 in shake flasks).
Awano et al.
[42][44] investigated the enzyme having CD activity in
E. coli using native-PAGE and CD activity staining. An analysis with gene-disrupted mutants showed that the tryptophanase (TNase) encoded by
tnaA and cystathionine β-lyase (CBL) encoded by
metC catalyze the degradation of L-cysteine in
E. coli.
The transcriptional activator CysB, belonging to the LysR family, induces the transcription of genes in the sulfate pathway
[43][46]. Kawano et al.
[43][46] found a most plausible sequence for the CysB-binding motif upstream of the
yciW gene. YciW encodes a soluble protein of 401 amino acids, although its function and structural information are still unknown. The CysB-binding motif is located at the −35 region of the predicted promotor, which is similar to that of
cysJ coding for a sulfite reductase. The
yciW-disrupted strain exhibited a much higher sensitivity to L-cysteine than the native strain. This indicates that
yciW confers tolerance to cysteine in
E. coli cells, and the authors suggest that
yciW may convert L-cysteine to L-methionine or glutathione. Based on the deduced amino acid sequence,
yciW is predicted to encode an oxidoreductase-like protein. YciW is also involved in the degradation of intracellular L-cysteine, leading to its detoxification. However, further investigation is needed at the protein level
[43][46].
L-cysteine desulfidases are alternative L-cysteine-degrading enzymes that are less well characterized as compared to L-cysteine desulfhydrases. The activity of this enzyme was first observed in
Methanocaldococcus jannaschii [44][47].
2.3. Regulation of L-Cysteine Transport
The bacterial production of metabolites depends on the equilibrium between secretion and reconsumption. High concentrations of L-cysteine have been reported to be toxic and inhibitory for microbial cells
[3]. In
E. coli, concentrations of 1 mM L-cysteine were enough to cause growth limitations
[43][46]. Therefore, microorganisms have an exporter for L-cysteine within the L-cysteine regulon in order to avoid product-associated cytotoxicity.
Daßler et al.
[45][51] identified the transporter protein YdeD in
E. coli, which is reported to be one of the main facilitators involved in the export of L-cysteine. The corresponding gene encoding for the transporter protein involves an open reading frame coding for 299 amino acids. The substrate range of the YdeD protein is wide; therefore, it secretes other metabolites along with L-cysteine.
CydDC is an ATP-binding transporter reported to be involved in L-cysteine export from the cytoplasm to the periplasm in
E. coli [46][53]. In a study, CydDC was compared to the gene product of
ydeD and was found to more effectively remove high L-cysteine concentrations in intracellular compartments than
ydeD. In shake flask experiments at 37 °C,
E. coli mutants with deletions of
cydDC accumulated 1.6 times higher L-cysteine concentrations than the wild type, while mutants with
ydeD deletions accumulated 1.3 times higher L-cysteine concentrations. A double-deletion
E. coli mutant of both
cydDC and
ydeD accumulated 2.2 times more L-cysteine than the wild type.
[46][53]
yfiK was discovered to be a gene that augmented L-cysteine production when it was overexpressed in an
E. coli production strain
[47][52]. The gene product is an integral membrane protein with about six predicted transmembrane helices and belongs to the resistance to homoserine/threonine (RhtB) family of export proteins. YfiK overproduction from a plasmid leads to the drastic and parallel secretion of O-acetyl serine and L-cysteine into the medium, but only when the
E. coli strain possesses an L-serine transacetylase that is feedback insensitive to L-cysteine. Externally provided, OAS obviated this requirement for L-cysteine secretion, both in the
yfiK-carrying transformant and in the wild type. A Δ
yfiK mutant did not show any phenotype, and this mutant exported OAS and L-cysteine when transformed with a plasmid carrying
ydeD, a previously characterized, alternate OAS/L-cysteine exporter. Since a
ydeD–yfiK double mutant showed the same pattern, it appears that YfiK and YdeD act independently. The necessity for the cell to regulate the size of the internal pool of OAS via the synthesis of exporter proteins could be connected to the fact that this compound inhibits growth when supplied externally. The overexpression of either
ydeD or
yfiK leads to alleviation of this inhibition parallel by an increased resistance to azaserine, which is an analog of OAS
[47][52].
2.4. Limitations of the Metabolic Engineering Approaches
Liu et al.
[13] developed an integrated approach, which used many of the previously presented metabolic engineering strategies in tandem. They overexpressed PSERT, PSL, and a feedback-insensitive version of PGDH, disrupted genes for the degradation of L-serine and L-cysteine, and overexpressed the L-cysteine exporter YdeD. Even though this strategy showed a cumulative improvement in the L-cysteine concentration, the final yield of L-cysteine on glucose was about 4.7% (
w/
w). This means that most of the fed substrate was not directly utilized for the L-cysteine production. Nonetheless, the authors indicate that this is the highest yield on glucose found in the literature at the time of its publication.
This exemplifies one of the major challenges of the metabolic engineering approaches. Most of the methods described above are applied directly on the L-cysteine synthesis pathway, which has been extensively characterized so that the bottlenecks are extensively identified. However, most metabolic pathways are thoroughly interconnected with regulation interactions that have not been yet characterized
[48][59]. These interactions must then first be mapped out in order to identify further bottlenecks that can be tackled through rational metabolic engineering methods
[48][59].
3. Process Engineering Aspects
3.1. Sulfur Assimilation Pathways in Bacteria
E. coli has two sulfur assimilation pathways for L-cysteine biosynthesis (
Figure 2). One is the sulfate (SO
42−) pathway, which consumes two molecules of ATP and four molecules of NADPH as a reducing power to produce L-cysteine from sulfate. The other is the thiosulfate (S
2O
32−) pathway, which spends only one molecule of NADPH from thiosulfate
[49][63]. Thiosulfate is receiving attention as a sulfur source due to its advantageous effects on bacterial growth and L-cysteine production in comparison to sulfate
[50][64].
Both sulfur assimilation pathways utilize OAS as a carbon skeleton to which they may incorporate sulfur. In the thiosulfate pathway, O-acetyl-L-serine sulfhydrylase B (CysM) catalyzes the conversion of OAS and thiosulfate into S-sulfocysteine, which is reduced into L-cysteine and sulfite by glutaredoxin (Grx1), and the glutaredoxin-like protein NrdH
[49][51][63,65]. In comparison, sulfate is first reduced to sulfide in a four-step reaction catalyzed in subsequent order by sulfate adenylyltransferase (CysDN), adenylylsulfate kinase (CysC), 3′-phospho-adenylylsulfate reductase (CysH), and sulfite reductase (CysIJ). The subsequent incorporation of sulfide into OAS is enabled by O-acetyl-L-serine sulfhydrylase A (CysK)
[52][60].
There is strong evidence suggesting that
E. coli may possess an alternative pathway for assimilating thiosulfate, independent of the conventional pathway through the CysM enzyme
[50][64]. This evidence comes from an experiment where ∆CysM
E. coli cells were grown using thiosulfate as the only sulfur source. The ∆CysM cells showed a slower growth compared to the wild-type cells. However, when L-cystine was added to the growth media, the impaired growth was restored, indicating a deficiency in L-cysteine. The growth rate in the growth media without L-cystine was then increased by overexpressing thiosulfate sulfurtransferase (GlpE).
3.2. Glycerol as Carbon Source
Although glucose remains the predominant carbon source for L-cysteine biosynthesis, glycerol has gained increasing attention as an alternative carbon source due to its abundant availability as a by-product in biodiesel production
[53][66]. Utilizing glycerol as a carbon source is crucial for reducing the costs of bioproduction and improving resource utilization. Moreover, the metabolic pathway from glycerol to L-cysteine is shorter and exhibits better carbon economy compared to glucose
[54][67].
Significant progress has been made towards the bioproduction of L-cysteine from glycerol, as its essential precursor, L-serine, has already been produced effectively using glycerol
[54][67]. The highest recorded concentration of L-cysteine from glycerol achieved so far is 313.4 mg L
−1 with
E. coli in shake flasks
[54][67]. First, L-serine production was enhanced by making use of an L-serine biosensor based on the transcriptional regulator NCgl0581 for coupling L-serine biosynthesis to the growth rate of
E. coli and using adaptive laboratory evolution (ALE).
3.3. Fed-Batch Processes in Stirred-Tank Bioreactors
Up to this point, most research presented had been carried out as simple batch processes in shake flasks, which allow for easier preparation and higher throughput than the scalable stirred-tank bioreactor. Investigations in fed-batch operated and fully controlled stirred-tank bioreactors using the observations provided by the studies in shake flasks and optimizing the reaction conditions may lead to an L-cysteine production scenario that is more in line with the expectations of the industry.
Liu et al.
[13] utilized a rational approach which combined previously discussed metabolic engineering approaches to enhance the L-cysteine production of the
E. coli strain JM109, including the overexpression of the enzymes in the L-serine biosynthetic pathway, feedback-insensitive phosphoglycerate dehydrogenase and serine acetyltransferase, the ydeD exporter, and the deletion of L-cysteine and serine deaminases. These modifications lead to a total L-cysteine production in shake flasks of 600 mg L
−1. They then cultivated this modified strain in a 5 L stirred-tank reactor in a fed-batch operation, where a glucose solution was fed constantly into the reactor, but thiosulfate (the sulfur source) was only supplied at the beginning of the process. This led to an increase in the maximal achieved L-cysteine concentration to 5.1 g L
−1.
3.4. Purification of L-Cysteine from the Fermentation Broth
L-cysteine, due to its high solubility in water and the vulnerability of its thiol group to oxidation, presents challenges in its purification for industrial applications. Several approaches have been proposed to address this issue
[55][62].
Most approaches follow a two-stage process, which involves converting L-cysteine into L-cystine by treating the clarified solution with an oxidizing agent. The reduced solubility and increased chemical stability of L-cystine allow for its purification through crystallization. The resulting crystals are then dissolved in water and reduced back to L-cysteine using electrolysis
[56][71].
More recently, Wacker AG (Munich, Germany) has developed a method for the direct purification of L-cysteine from a clarified fermentation broth
[55][62]. This approach selectively isolates L-cysteine while eliminating its derivatives, emphasizing the importance of preventing the oxidation of L-cysteine to L-cystine. The oxidation of L-cysteine’s sulfhydryl (SH) groups by oxygen, the primary oxidizing agent present in fermentation broths, is particularly favored under alkaline conditions.
4. Conclusions
In general, the most widely researched microorganism for L-cysteine bioproduction is
E. coli. Nonetheless, other production hosts like
C. glutamicum and
P. ananatis have been the focus of strain optimization, with promising improvements in L-cysteine production.
The combination of three metabolic engineering strategies has been successful, irrespective of the production host: The first one is enhancing the L-cysteine biosynthesis by overexpressing the genes encoding for feedback-insensitive L-serine O-acetyltransferase (SAT) and O-acetyl-L-serine sulfhydrylase. In
E. coli, the feedback inhibition of SAT was bypassed through the introduction of an SAT from
Arabidopsis thaliana.
C. glutamicum showed a high L-cysteine production with the homologous overexpression of the
cysE and
serA genes.
The second metabolic engineering strategy is to reduce L-cysteine degradation by the microorganisms. Therefore, the L-cysteine desulfhydrase gene
aecD was deleted in
C. glutamicum, which increased the production of L-cysteine. In
E. coli, the gene products of
tnaA and
metC were identified to catalyze the degradation of L-cysteine, and after the deletion of these genes, the L-cysteine production was improved.
The third metabolic engineering strategy is to enhance the export of L-cysteine and weaken the import. As L-cysteine is cytotoxic, it has to be transported out of the cell. In
E. coli, two membrane proteins called YdeD and YfiK are responsible for the export of L-cysteine. But by overexpression of
ydeD, OAS is simultaneously secreted, which causes the L-cysteine regulon activation to stop. In contrast, the gene product of
yfiK enables L-cysteine export without depleting the intracellular OAS concentrations. In addition, the gene product of
yeaN is a recently discovered L-cysteine uptake protein found in
E. coli, which may be a promising target to avoid the reconsumption of L-cysteine by the cells.
Taking into account the process engineering aspects beyond metabolic engineering of the L-cysteine biosynthesis pathway enables economic L-cysteine production by fermentation. Emphasizing energy-efficient sulfur pathways, selecting economically viable carbon sources, designing scalable fed-batch processes with individual feedings of carbon and sulfur sources, and implementing efficient purification techniques are important for the production of L-cysteine on an industrial scale. The utilization of thiosulfate instead of sulfates requires fewer reduction equivalents, and thus, enhances L-cysteine production rates. Even though the highest L-cysteine concentrations are still obtained with glucose as a carbon source, microbial L-cysteine production with glycerol has been shown. Glycerol is abundant as a by-product of the biodiesel industry, and thus, its low cost might improve the industrial-scale L-cysteine production in the future.