DNA methylation is a fundamental mechanism of epigenetic control in cells and its dysregulation is strongly implicated in cancer development. Cancers possess an extensively hypomethylated genome with focal regions of hypermethylation at CPG islands. Due to the highly conserved nature of cancer-specific methylation, its detection in cell-free DNA in plasma using liquid biopsies constitutes an area of interest in biomarker research. The advent of next-generation sequencing and newer computational technologies have allowed for the development of diagnostic and prognostic biomarkers that utilize methylation profiling to diagnose disease and stratify risk. Methylome-based predictive biomarkers can determine the response to anti-cancer therapy. An additional emerging application of these biomarkers is in minimal residual disease monitoring. Several key challenges need to be addressed before cfDNA-based methylation biomarkers become fully integrated into practice. The first relates to the biology and stability of cfDNA. The second concerns the clinical validity and generalizability of methylation-based assays, many of which are cancer type-specific. The third involves their practicability, which is a stumbling block for translating technologies from bench to clinic.
- DNA methylation
- liquid biopsy
- biomarkers
- cell-free DNA
- 5-methylcytosine
1. Introduction
2. Biology of DNA Methylation in Malignancy
2.1. Aberrant DNA Methylation of CpG Islands Are Seen in Cancer
More than half of all human promoters overlap with 1–2 kb CG-rich regions, referred to as CpG islands (CGIs). Nearly 95% of CGI promoter regions are unmethylated in normal tissues. While CGIs are preferentially found near promoters and transcription start sites (TSS), they may also appear within gene regulatory elements, gene bodies, and intergenic regions [1]. Cancers typically display focal regions of tumor-specific hypermethylation, especially at CGIs, against the backdrop of global hypomethylation that impacts gene expression and carcinogenesis [2][3][4].2.2. Gene Promoter Hypermethylation Leads to Silencing of Tumor Suppressor Genes
Hypermethylation of transcriptional regulatory regions of tumor suppressor genes (TSGs) results in their silencing and the abrogation of their anti-tumor function [2][5]. First described in colorectal cancer (CRC), the CpG island methylator phenotype (CIMP) that is characterized by the expansive TSG promoter silencing has since been documented in other solid malignancies [6]. DNA hypermethylation was observed in promoters of classic TSGs such as retinoblastoma transcriptional corepressor 1 (RB1) and cell cycle regulators CDKN2A, among many others [1][2].2.3. Methylation Can Lead to Epigenetic Activation of Oncogenic Pathways
Hypomethylation of oncogene promoters typically results in their expression. Oncogene products dysregulate cell proliferation, migration, and invasion. This results in elevated metastatic potential, increased stemness in cells, or immune evasion [7]. For example, BCL6 acts as a transcriptional regulator that represses tumor suppressors thereby favoring cell proliferation and survival [8]. Expression of BCL6 is epigenetically regulated and knockdown of UHRF1, an upstream regulator, results in loss of BCL6 gene promoter methylation thereby favoring its expression. Notably, BCL6 was observed to be overexpressed in exhausted T-cells and may play a role in tumor immune escape [9]. In addition to promoters, hypomethylation of other gene regulatory regions, such as enhancers, was shown to correlate with the expression of cancer-specific genes and pathways across multiple cancers [10][11]. In contrast to promoter hypermethylation, gene body hypermethylation is posited to correlate positively with gene expression [1][12][13]. As an example, the homeobox superfamily of transcription factors is essential to the regulation of cell growth and differentiation. These genes have high CpG density, which makes their expression susceptible to epigenetic control [14]. In relation to this, Su et al. [15] reported that gene-body hypermethylation results in the overexpression of approximately 43% of known homeobox genes, many of which are associated with oncogenesis.2.4. Global DNA Hypomethylation Leads to Chromosomal Instability
DNA methylation plays a critical role in maintaining genomic integrity by regulating the transcription of constitutive heterochromatin elements, sub-telomeric regions, and genomic repeats [16][17]. Expansive hypomethylation is common in cancer and may involve regions as large as ten megabases in size, which can render the cancer genome susceptible to genomic instability. Moreover, increased gene transcription from loss of gene promoter methylation results in the formation of DNA-RNA R-loops that can cause replication stress and DNA damage—further exacerbating genome instability [16][18]. Reduced methylation destabilizes heterochromatin at pericentromeric regions and is associated with loss of tumor suppression and oncogenesis in gastrointestinal cancers and sarcomas [16][17]. Telomere length is affected by methylation of sub-telomeric regions, and its dysregulation is considered a cancer hallmark [16][19][20].2.5. DNA Methylation Influences Gene Expression beyond Promoter Silencing
Alternative mRNA splicing is responsible for transcriptional diversity in cells [21][22]. During splicing, non-coding introns are removed, and coding exons are ligated to form mature mRNA at specific positions called splice sites. Recognition of introns and exons by the spliceosome machinery is critical for the correct execution of this process [21]. DNA methylation modifies the accessibility of exonic nucleosomes to RNA Polymerase II, which demonstrates the regulatory role it plays in splicing [21][22]. Chen and Elnitski [23] showed that methylation-correlated isoforms affect the functional protein domains of gene products. Moreover, these gene sets were enriched for oncogenes, tumor suppressors, and cancer-related pathways across multiple cancers [23].2.6. Methylation Can Influence the Non-Coding Genome, Leading to Cancer
DNA hypomethylation led to the expression of genomic repeat elements and transposable elements that interfered with chromosomal integrity and drove oncogene expression in a process called onco-exaptation [16][17]. Jang et al. [24] reported RNA-seq data from 7769 tumors across 15 cancer types from the TCGA. In this cohort, the expression of 106 oncogenes was regulated by onco-exaptation in nearly half (49.7%) of tumors. To validate this relationship, the authors performed in vitro induction of DNA methylation on the promoter of the short-interspersed nuclear element (SINE) AluJb by CRISPR. This resulted in a 20–30% increase in methylation and consequently a 40% reduction in the expression of the oncogene LIN28B. In addition to onco-exaptation, transposon-associated dsRNA-dependent paracrine signaling had been found to be favorable to tumor growth in breast, lung, and pancreatic cancer [25][26]. The study of non-coding RNA is a growing area of interest owing to the emerging roles that it plays in disease [27][28]. The interaction between non-coding RNA (ncRNA) and DNA methylation is complex. Few studies have comprehensively examined if these non-coding regions are methylated, with most investigations focusing on a specific type of ncRNA called long non-coding RNA (lncRNA). Several comprehensive reviews discuss the association between DNA methylation and lncRNA expression in cancer [29][30]. In contrast to lncRNAs, other types of noncoding RNA have not been examined as extensively in the context of DNA methylation. This underscores the complexity of the relationship between ncRNA and DNA methylation in cancer [31].2.7. Role of DNA Methylation in the Tumor Microenvironment
Methylation dynamics play a key role in contextualizing the tumor microenvironment (TME). To illustrate, TET2 demethylates promoters of genes encoding cytokines and transcription factors that influence CD4+ T-cell fate. Expression of the cytokine IL-4 favors differentiation into Th2 cells while expression of the transcriptional regulator FOXP3 expression leads to differentiation into tissue regulatory T-cells (Treg) [32]. In CD8+ cells, active demethylation of gene enhancer regions by TET2 facilitates differentiation to T-effector cells. Conversely, the conditional loss of TET activity was shown to drive a shift towards a memory T-cell fate [32][33][34]. Meanwhile, DNMT3A-directed de novo methylation of the promoter of T-cell-specific transcription factor 7 (Tcf7) results in the suppression of memory T-cell differentiation and supports differentiation into effector subtypes [33][34]. As with T-cells, myeloid differentiation is epigenetically influenced by DNA methylation. TET2 acts in concert with IL-4 and STAT6 to promote the expression of ITGB2, which is a cell adhesion molecule important in monocyte-to-dendritic cell differentiation and function. Dendritic cells have a central role in antigen presentation and T-cell activation within the TME [32]. Myeloid-derived suppressor cells (MDSC) are key immune regulators in the TME that serve a pro-tumorigenic function by promoting immune tolerance to cancer. An increase in DNMT3A expression in MDSCs in the presence of Prostaglandin E2 results in a gain of DNA methylation and silencing of immunogenicity-associated genes (e.g., FAS, RUNX1, and S1PR4) [35][36]. Finally, macrophage polarization towards either the activated (M1) phenotype or the inhibitory (M2) phenotype is also subject to the influence of DNA methylation. Express of DNMT3b and DNMT1 are associated with M1-like macrophage polarization through its effect on PPAR γ1 [37]. Meanwhile, siRNA knockdown of the DNMT3B gene increases M2 marker expression in macrophages. A similar increase in M2 markers was seen following the treatment of M1 macrophages with DNMT inhibitors (DNMTi) [37][38][39]. The development of immune escape mechanisms that bar cytotoxic effector cells from infiltrating the TME is key to tumor progression [20]. DNA methylation also plays a part in this process. DNMT1 activity was shown to disturb the trafficking of CD8+ T-cells into the TME of a murine ovarian cancer cell model by repressing the tumor production of CXCL9 and CXCL10 [32]. In addition, tumors use DNA methylation to negatively regulate chemokines such as CCL5 and CCL2, which are essential in T-cell and macrophage chemotaxis. Epigenetic silencing of the CCL2 gene by methylation was found to diminish macrophage infiltration and promote disease progression in small-cell lung cancer (SCLC) [40][41][42]. As described previously, DNA methylation also plays a role in the activation of inhibitory immune cells such as MDSC that modulate a pro-tumor TME. Interestingly, MDSC infiltration can be reduced by treatment with DNMTi [43]. Investigations into the mechanisms behind effector T-cell exhaustion revealed the contribution of multiple epigenetic mechanisms including DNA methylation. De novo methylation by DNMT3A established a stable exhausted state and its deletion in CAR T-cells as shown by Prinzing et al. [44] led to a more anti-tumor state. Moreover, DNA methylation profiling of exhausted tumor-infiltrating lymphocytes (TIL) identified promoter hypomethylation of T-cell exhaustion markers PD-1 and HAVCR2. Binding sites of exhaustion-associated transcription factors such as NR4A1 were also seen to be hypomethylated, implying that activation of the T-cell inhibitory programming was governed, at least in part, by methylation [45]. Taken together, these highlight the complex roles DNA methylation plays in the tumor TME.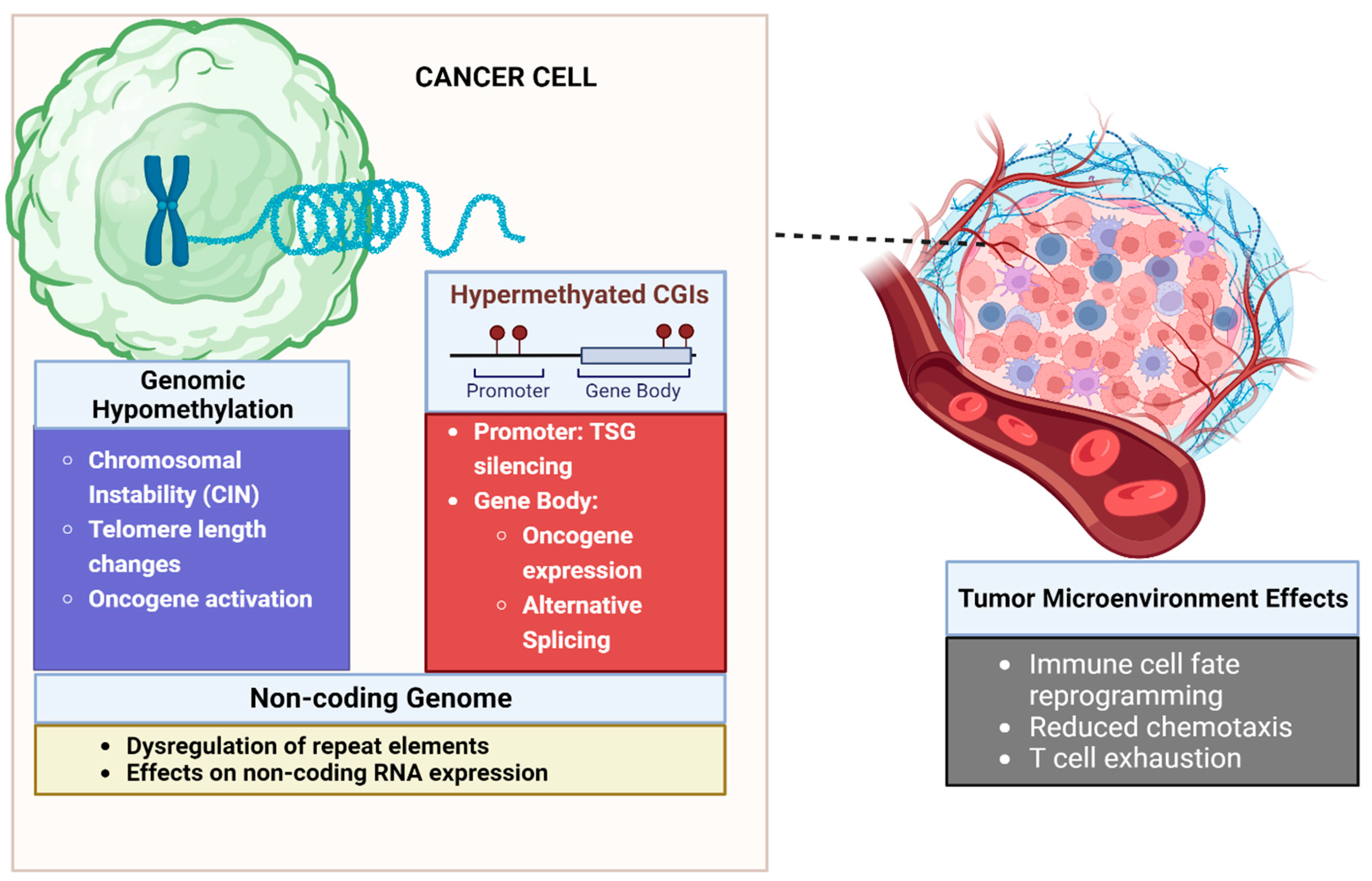
3. Cell-Free Tumor Methylome Recapitulates the Cancer Epigenome
Cell-free DNA (cfDNA) refers to DNA fragments in the noncellular component of blood. Although it has been an area of great interest over the last decade, Mandel and Métais were the first to demonstrate DNA in the blood of both healthy and diseased patients in 1948 [46]. cfDNA arises from cells through necrosis and apoptosis, but also enters the circulation via active secretion [46][47]. Circulating DNA from normal cells is found in low concentrations in plasma (~10–15 ng/mL) but can increase due to acute stressors like disease and injury [48]. Notably, it was shown that levels of cfDNA in individuals with cancer are higher in comparison with their counterparts without cancer [48]. The component of cfDNA that originates from tumors is referred to as circulating tumor DNA (ctDNA) and represents a smaller fraction of overall cfDNA [49][50]. These circulating tumor fragments are shorter than DNA from non-cancer cells and range in size from ~140 to 200 bp in length, corresponding to the size of nucleosome-associated DNA [46][51]. The ctDNA fraction in plasma varies but increases in association with disease type and burden. Advanced disease and high-shedding tumors such as SCLC and prostate cancer may have fractions > 20% of total cfDNA [52][53][54][55]. Conversely, reduced ctDNA fractions in the blood are detectable in early-stage disease or following effective anticancer therapy [48][52][54][56]. In addition to plasma, ctDNA can be obtained from other biofluids such as cerebrospinal fluid, saliva, pleural effusions, urine, ascites, and stool representing multiple sources of nucleic acid for study [48]. Depending on the source, ctDNA size may vary because of disparate conditions within the respective biological compartments from which the nucleic acids are sourced. For instance, shorter ctDNA fragments may be observed in urine compared to plasma owing to the greater nuclease activity in the former [46]. While the term liquid biopsy is most often used to refer to the analysis of cfDNA from peripheral blood, it also pertains to the isolation and study of tumor-derived material from other bodily fluids. Liquid biopsies using standard venipuncture afford several distinct advantages over conventional tissue sampling. First, their minimally invasive nature allows for evaluation of the disease, particularly in cases where tumor access is difficult, unsafe, or ethically challenging, especially for repeated longitudinal sampling [48]. Moreover, in addition to ctDNA, circulating tumor microRNA, extracellular vesicles, tumor-associated proteins, and circulating tumor cells (CTCs) can also be sampled at the point of care by this approach [57]. From a testing standpoint, utilizing pooled cfDNA obtained from liquid biopsies to determine a positive signal—such as the presence of cancer in a patient—avoids dependence on specific fragments and thereby improves sensitivity. This is especially important when dealing with scenarios wherein the amount of input DNA is constrained, and adequate representation of individual DNA fragments cannot be guaranteed [58]. Another advantage of liquid biopsies is their ability to better recapitulate the molecular heterogeneity harbored by multiple distinct clonal populations that collectively shed cfDNA into the blood. This is because the molecular diversity of disease is not as easily captured by needle aspirates of single tumor sites and multiple needle passes to sample different sites may be difficult or excessively invasive [48]. Finally, from a therapeutic perspective, this technique allows for longitudinal sampling across successive time points providing opportunities for studying tumor evolution resulting from treatment [48][50]. Early applications of liquid biopsy technology involve genotyping and mutation detection, but indications for its use have since expanded to include epigenetic analyses, including the study of ctDNA methylation [50][59][60][61]. The study of ctDNA methylation in turn has several advantages. First, cancer-specific methylation changes occur earlier in tumorigenesis and represent an opportunity for early disease detection and diagnosis [49][62]. Moreover, combining methylation with orthogonal methods such as mutation detection and fragmentomics to profile ctDNA further enhances the discriminative power of such tests by interrogating biologic features that are characteristic of tumor-derived DNA [63]. Second, methylation patterns in ctDNA are consistent with their tissue-of-origin, allowing for the location of the source of a cancer, especially those with an unknown primary site [60][64]. Third, tumor cells tend to exhibit more homogenous DNA methylation changes versus gene mutations that tend to exhibit greater intra-tumoral heterogeneity [59]. The consistency of the methylation signal opens the door for evaluating tumor evolution, especially after being subjected to the selective pressure of anti-cancer therapy. In the context of treatment resistance, examining changes in differentially methylated regions (DMR) in ctDNA obtained from longitudinal blood draws can shed light on epigenetic mechanisms that underpin treatment resistance. Various bisulfite conversion (e.g., quantitative methylation-specific PCR and cfMethyl-seq) and antibody-enrichment-based (e.g., cfMeDIP-seq) platforms for the study of ctDNA methylation have been developed in the last decade. The details of these assays are beyond the scope of this research but were extensively discussed elsewhere [65][66][67][68].4. Applications of cfDNA Methylation as Biomarker in Cancer
4.1. Methylation as a Diagnostic Biomarker
The concordance between tumor and cell-free cancer methylomes means that it can be leveraged as a tool for cancer diagnosis. Assays that employ ctDNA methylation for this application vary in their approaches. Some use targeted detection of methylated sites in specific genes such as SEPT9 [69][70], SHOX2 [71], PTGER4 [71], and SDC2 [72] while others profile more extensive swathes of the genome. Classifiers based on more extensive DNA methylation profiles to detect cancers via plasma-based liquid biopsies were also developed. In this regard, the most significant application of ctDNA methylome profiling is in multicancer early detection (MCED). The importance of this application lies in the fact that early diagnosis of disease allows for the administration of effective, curative treatment to optimize patient outcomes [73]. A second platform, this time that leveraged cfMeDIP-seq, an antibody-based, non-degradative genome-wide DNA methylation enrichment assay was being developed for use in MCED. Validation of this test was performed on a cohort of 4332 consisting of untreated patients across eight different tumor types—bladder, breast, CRC, head, and neck (HNSCC), lung, ovarian, prostate, and renal cell (RCC) cancers, and a set of age and sex-matched controls [74]. The test performed well with an AUROC of 0.94 (95% CI: 0.93, 0.96) across all tumors and was robust in each individual tumor type including low cfDNA shedding tumors—bladder, breast, prostate, and RCC—with an AUROC 0.92 (0.91, 0.94) [74]. The high detection of low-shedding disease was particularly noteworthy since early-stage cancers with low tumor burden released small amounts of cfDNA into the circulation and were an important hurdle to overcome for viable MCED tests.4.2. Methylation as a Prognostic Biomarker of Patient Outcomes
As a ubiquitous mode of epigenetic regulation, DNA methylation of specific genes provides value in prognosticating outcomes in cancer. Huang et al. [75] utilized bisulfite conversion-based real-time PCR to determine the methylation status of the SEPT9 gene (mSEPT9) in the plasma of 144 preoperative CRC patients. Patients who were mSEPT9+ had lower disease-free survival (DFS) rates than those who were mSEPT9− (two-year DFS: 52.1% vs. 73.9%, p = 0.014), Moreover, mSEPT9 was an independent predictor of prognosis (HR = 2.741, p = 0.009) in multivariate regression analysis [75]. Of interest, a 2021 systematic review by Hier et al. [76] identified over 100 different genes regulated by methylation that carried prognostic significance. Apart from protein-coding genes, the loss of methylation of repeat elements was significant in early carcinogenesis and correlated with poor prognostic outcomes [77][78]. Recognizing that the tumor methylation signal was distinct from that of the non-cancer genome, Ul Haq et al. [79] performed a whole genome approach to the assessment of the cell-free methylome of SCLC using cfMeDIP-seq. The authors utilized an in silico approach using paired peripheral blood leukocytes to subtract non-cancer noise from cfDNA methylation data similar to previous studies [63]. This approach identified two biologically distinct methylation-defined SCLC prognostic groups with differences in overall survival [79]. Validation of ctDNA methylation as a prognostic biomarker in HNSCC and RCC was performed and data from these studies were presented in abstract form at the ESMO 2023 conference [80][81].4.3. Methylation as a Predictive Biomarker of Treatment Response
Methylation of the O6-alkylguanine DNA alkyltransferase (MGMT) promoter had been a known predictor of response to the alkylator temozolomide in glioma [57]. The promoter methylation status of this gene could be assessed using ctDNA obtained from blood or cerebrospinal fluid via targeted, methylation-specific PCR and was found to be highly concordant with tumor tissue [82]. Meanwhile, the expression of the DNA/RNA helicase Schlafen 11 (SLFN11) predicted sensitivity to DNA-damaging alkylating agents and PARP inhibitors [83][84][85][86]. The SLFN11 gene was observed to be under epigenetic control and methylation of its promoter in ctDNA predicted resistance to therapy in ovarian cancer with the prior mentioned agents [87]. Hypermethylation of the promoter of the MLH1 gene represented a principal epigenetic mechanism that led to the MSI-H/dMMR state. Tumors with deficient mismatch repair possessed a higher burden of somatic mutations, higher infiltrating T-cell counts, and showed increased PD-L1 positivity—all these predispose MSI-H/dMMR cancers to respond to immune checkpoint inhibitor (ICI) therapy [88][89]. Wang et al. [90] described a liquid biopsy-based approach to the detection of MLH1 promoter methylation in CRC using methylation-sensitive restriction enzyme PCR. This assay had an AUROC value of 0.965 (95% CI: 0.94–0.99). The sensitivity and specificity of the assay were 78% and 100%, respectively (95% CI: 0.45–0.95). Beyond individual genes, methylation-profile-based scoring also predicted anti-cancer therapy response. In an exploratory study from the phase 2 SWOG S1314 clinical trial, pre-treatment cfDNA from 72 patients was used to generate a classifier methylation-based response score (mR-score), which could predict pathologic response to neoadjuvant chemotherapy in operable, muscle-invasive bladder cancer (MIBC). The model demonstrated an AUROC of 0.636 (95% CI: 0.498–0.773) for baseline samples. Moreover, mR-scores correlated with pathologic response with complete responders demonstrating the lowest scores in the cohort and non-responders showing the highest mR-scores. Notably, this same classifier showed better performance when applied to on-treatment samples (AUROC of 0.720, 95% CI: 0.582–0.857) [91]. Methylation-profile-based scoring was also used to predict therapy response in ICI.4.4. Minimal Residual Disease
Effective oncologic therapy reduces tumor burden by >2–3 log10 which corresponds to a tumor kill of >99%. Unfortunately, malignant cells may persist in patients who achieve exquisite treatment responses and represent niduses for disease relapse. These recalcitrant cancer cells, referred to as minimal residual disease (MRD), remain a key challenge in the treatment of cancer [92]. To guarantee the durability of remission following curative-intent therapy, increasingly sensitive methods for post-treatment surveillance are necessary [50]. Knowing that recurrent tumors detectable by conventional imaging contain >106 cancer cells, the likelihood of successful treatment becomes less likely once it is demonstrable by diagnostic scans [93]. Detection of ctDNA by liquid biopsies of different biofluids, such as plasma and urine, represents an avenue for earlier detection of recurrent disease thereby facilitating cancer interception, earlier intervention, and more optimal treatment outcomes [94][95][96][97][98][99].References
- Wang, Q.; Xiong, F.; Wu, G.; Liu, W.; Chen, J.; Wang, B.; Chen, Y. Gene Body Methylation in Cancer: Molecular Mechanisms and Clinical Applications. Clin. Epigenetics 2022, 14, 154.
- Nishiyama, A.; Nakanishi, M. Navigating the DNA Methylation Landscape of Cancer. Trends Genet. 2021, 37, 1012–1027.
- Yokoyama, T.; Takehara, K.; Sugimoto, N.; Kaneko, K.; Fujimoto, E.; Okazawa-Sakai, M.; Okame, S.; Shiroyama, Y.; Yokoyama, T.; Teramoto, N.; et al. Lynch Syndrome-Associated Endometrial Carcinoma with MLH1 rmline Mutation and MLH1 Promoter Hypermethylation: A Case Report and Literature Review. BMC Cancer 2018, 18, 576.
- Joo, J.E.; Mahmood, K.; Walker, R.; Georgeson, P.; Candiloro, I.; Clendenning, M.; Como, J.; Joseland, S.; Preston, S.; Graversen, L.; et al. Identifying Primary and Secondary MLH1 Epimutation Carriers Displaying Low-Level Constitutional MLH1 Methylation Using Droplet Digital PCR and Genome-Wide DNA Methylation Profiling of Colorectal Cancers. Clin. Epigenetics 2023, 15, 95.
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505.
- Yates, J.; Boeva, V. Deciphering the Etiology and Role in Oncogenic Transformation of the CpG Island Methylator Phenotype: A Pan-Cancer Analysis. Brief. Bioinform. 2022, 23, bbab610.
- Romero-Garcia, S.; Prado-Garcia, H.; Carlos-Reyes, A. Role of DNA Methylation in the Resistance to Therapy in Solid Tumors. Front. Oncol. 2020, 10, 1152.
- McLachlan, T.; Matthews, W.C.; Jackson, E.R.; Staudt, D.E.; Douglas, A.M.; Findlay, I.J.; Persson, M.L.; Duchatel, R.J.; Mannan, A.; Germon, Z.P.; et al. B-Cell Lymphoma 6 (BCL6): From Master Regulator of Humoral Immunity to Oncogenic Driver in Pediatric Cancers. Mol. Cancer Res. 2022, 20, 1711–1723.
- Zhang, C.; Sheng, Q.; Zhao, N.; Huang, S.; Zhao, Y. DNA Hypomethylation Mediates Immune Response in Pan-Cancer. Epigenetics 2023, 18, 2192894.
- Ankill, J.; Aure, M.R.; Bjørklund, S.; Langberg, S.; Kristensen, V.N.; Vitelli, V.; Tekpli, X.; Fleischer, T. Epigenetic Alterations at Distal Enhancers Are Linked to Proliferation in Human Breast Cancer. NAR Cancer 2022, 4, zcac008.
- Cho, J.-W.; Shim, H.S.; Lee, C.Y.; Park, S.Y.; Hong, M.H.; Lee, I.; Kim, H.R. The Importance of Enhancer Methylation for Epigenetic Regulation of Tumorigenesis in Squamous Lung Cancer. Exp. Mol. Med. 2022, 54, 12–22.
- Pongor, L.S.; Tlemsani, C.; Elloumi, F.; Arakawa, Y.; Jo, U.; Gross, J.M.; Mosavarpour, S.; Varma, S.; Kollipara, R.K.; Roper, N.; et al. Integrative Epigenomic Analyses of Small Cell Lung Cancer Cells Demonstrates the Clinical Translational Relevance of Gene Body Methylation. iScience 2022, 25, 105338.
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene Body Methylation Can Alter Gene Expression and Is a Therapeutic Target in Cancer. Cancer Cell 2014, 26, 577–590.
- Feng, Y.; Zhang, T.; Wang, Y.; Xie, M.; Ji, X.; Luo, X.; Huang, W.; Xia, L. Homeobox Genes in Cancers: From Carcinogenesis to Recent Therapeutic Intervention. Front. Oncol. 2021, 11, 770428.
- Su, J.; Huang, Y.-H.; Cui, X.; Wang, X.; Zhang, X.; Lei, Y.; Xu, J.; Lin, X.; Chen, K.; Lv, J.; et al. Homeobox Oncogene Activation by Pan-Cancer DNA Hypermethylation. Genome Biol. 2018, 19, 108.
- Besselink, N.; Keijer, J.; Vermeulen, C.; Boymans, S.; de Ridder, J.; van Hoeck, A.; Cuppen, E.; Kuijk, E. The Genome-Wide Mutational Consequences of DNA Hypomethylation. Sci. Rep. 2023, 13, 6874.
- Chen, R.; Ishak, C.A.; De Carvalho, D.D. Endogenous Retroelements and the Viral Mimicry Response in Cancer Therapy and Cellular Homeostasis. Cancer Discov. 2021, 11, 2707–2725.
- García-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618.
- Tire, B.; Ozturk, S. Potential Effects of Assisted Reproductive Technology on Telomere Length and Telomerase Activity in Human Oocytes and Early Embryos. J. Ovarian Res. 2023, 16, 130.
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46.
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; Caesar-Johnson, S.J.; Demchok, J.A.; Felau, I.; et al. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep. 2018, 23, 282–296.e4.
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and Mechanisms of Alternative Splicing in Cancer—Implications for Care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474.
- Chen, Y.-C.; Elnitski, L. Aberrant DNA Methylation Defines Isoform Usage in Cancer, with Functional Implications. PLoS Comput. Biol. 2019, 15, e1007095.
- Jang, H.S.; Shah, N.M.; Du, A.Y.; Dailey, Z.Z.; Pehrsson, E.C.; Godoy, P.M.; Zhang, D.; Li, D.; Xing, X.; Kim, S.; et al. Transposable Elements Drive Widespread Expression of Oncogenes in Human Cancers. Nat. Genet. 2019, 51, 611–617.
- Espinet, E.; Gu, Z.; Imbusch, C.D.; Giese, N.A.; Büscher, M.; Safavi, M.; Weisenburger, S.; Klein, C.; Vogel, V.; Falcone, M.; et al. Aggressive PDACs Show Hypomethylation of Repetitive Elements and the Execution of an Intrinsic IFN Program Linked to a Ductal Cell of Origin. Cancer Discov. 2021, 11, 638–659.
- Tavora, B.; Mederer, T.; Wessel, K.J.; Ruffing, S.; Sadjadi, M.; Missmahl, M.; Ostendorf, B.N.; Liu, X.; Kim, J.-Y.; Olsen, O.; et al. Tumoural Activation of TLR3–SLIT2 Axis in Endothelium Drives Metastasis. Nature 2020, 586, 299–304.
- Liu, S.J.; Dang, H.X.; Lim, D.A.; Feng, F.Y.; Maher, C.A. Long Noncoding RNAs in Cancer Metastasis. Nat. Rev. Cancer 2021, 21, 446–460.
- Bunch, H. Gene Regulation of Mammalian Long Non-Coding RNA. Mol. Genet. Genom. 2018, 293, 1–15.
- Al-Imam, M.J.; Hussein, U.A.-R.; Sead, F.F.; Faqri, A.M.A.; Mekkey, S.M.; Khazel, A.J.; Almashhadani, H.A. The Interactions between DNA Methylation Machinery and Long Non-Coding RNAs in Tumor Progression and Drug Resistance. DNA Repair 2023, 128, 103526.
- Sideris, N.; Dama, P.; Bayraktar, S.; Stiff, T.; Castellano, L. LncRNAs in Breast Cancer: A Link to Future Approaches. Cancer Gene Ther. 2022, 29, 1866–1877.
- Shen, S.; Chen, J.; Li, H.; Jiang, Y.; Wei, Y.; Zhang, R.; Zhao, Y.; Chen, F. Large-Scale Integration of the Non-Coding RNAs with DNA Methylation in Human Cancers. Cell Rep. 2023, 42, 112261.
- Zhong, F.; Lin, Y.; Zhao, L.; Yang, C.; Ye, Y.; Shen, Z. Reshaping the Tumour Immune Microenvironment in Solid Tumours via Tumour Cell and Immune Cell DNA Methylation: From Mechanisms to Therapeutics. Br. J. Cancer 2023, 129, 24–37.
- Sun, L.; Su, Y.; Jiao, A.; Wang, X.; Zhang, B. T Cells in Health and Disease. Signal Transduct. Target. Ther. 2023, 8, 235.
- Henning, A.N.; Roychoudhuri, R.; Restifo, N.P. Epigenetic Control of CD8+ T Cell Differentiation. Nat. Rev. Immunol. 2018, 18, 340–356.
- Rodríguez-Ubreva, J.; Català-Moll, F.; Obermajer, N.; Álvarez-Errico, D.; Ramirez, R.N.; Company, C.; Vento-Tormo, R.; Moreno-Bueno, G.; Edwards, R.P.; Mortazavi, A.; et al. Prostaglandin E2 Leads to the Acquisition of DNMT3A-Dependent Tolerogenic Functions in Human Myeloid-Derived Suppressor Cells. Cell Rep. 2017, 21, 154–167.
- Liu, R.; Zhao, E.; Yu, H.; Yuan, C.; Abbas, M.N.; Cui, H. Methylation across the Central Dogma in Health and Diseases: New Therapeutic Strategies. Signal Transduct. Target. Ther. 2023, 8, 310.
- Wang, X.; Cao, Q.; Yu, L.; Shi, H.; Xue, B.; Shi, H. Epigenetic Regulation of Macrophage Polarization and Inflammation by DNA Methylation in Obesity. JCI Insight 2016, 1, e87748.
- Chen, C.; Liu, T.; Tang, Y.; Luo, G.; Liang, G.; He, W. Epigenetic Regulation of Macrophage Polarization in Wound Healing. Burns Trauma 2023, 11, tkac057.
- Vadevoo, S.M.P.; Gunassekaran, G.R.; Yoo, J.D.; Kwon, T.-H.; Hur, K.; Chae, S.; Lee, B. Epigenetic Therapy Reprograms M2-Type Tumor-Associated Macrophages into an M1-like Phenotype by Upregulating MiR-7083-5p. Front. Immunol. 2022, 13, 976196.
- Dangaj, D.; Bruand, M.; Grimm, A.J.; Ronet, C.; Barras, D.; Duttagupta, P.A.; Lanitis, E.; Duraiswamy, J.; Tanyi, J.L.; Benencia, F.; et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 2019, 35, 885–900.e10.
- Zheng, Y.; Wang, Z.; Wei, S.; Liu, Z.; Chen, G. Epigenetic Silencing of Chemokine CCL2 Represses Macrophage Infiltration to Potentiate Tumor Development in Small Cell Lung Cancer. Cancer Lett. 2021, 499, 148–163.
- Khan, P.; Fatima, M.; Khan, M.A.; Batra, S.K.; Nasser, M.W. Emerging Role of Chemokines in Small Cell Lung Cancer: Road Signs for Metastasis, Heterogeneity, and Immune Response. Semin. Cancer Biol. 2022, 87, 117–126.
- Chen, X.; Pan, X.; Zhang, W.; Guo, H.; Cheng, S.; He, Q.; Yang, B.; Ding, L. Epigenetic Strategies Synergize with PD-L1/PD-1 Targeted Cancer Immunotherapies to Enhance Antitumor Responses. Acta Pharm. Sin. B 2020, 10, 723–733.
- Prinzing, B.; Zebley, C.C.; Petersen, C.T.; Fan, Y.; Anido, A.A.; Yi, Z.; Nguyen, P.; Houke, H.; Bell, M.; Haydar, D.; et al. Deleting DNMT3A in CAR T Cells Prevents Exhaustion and Enhances Antitumor Activity. Sci. Transl. Med. 2023, 13, eabh0272.
- Yang, R.; Cheng, S.; Luo, N.; Gao, R.; Yu, K.; Kang, B.; Wang, L.; Zhang, Q.; Fang, Q.; Zhang, L.; et al. Distinct Epigenetic Features of Tumor-Reactive CD8+ T Cells in Colorectal Cancer Patients Revealed by Genome-Wide DNA Methylation Analysis. Genome Biol. 2019, 21, 2.
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid Biopsies Come of Age: Towards Implementation of Circulating Tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238.
- Gaitsch, H.; Franklin, R.J.M.; Reich, D.S. Cell-Free DNA-Based Liquid Biopsies in Neurology. Brain 2023, 146, 1758–1774.
- Corcoran, R.B.; Chabner, B.A. Application of Cell-Free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765.
- Lau, B.T.; Almeda, A.; Schauer, M.; McNamara, M.; Bai, X.; Meng, Q.; Partha, M.; Grimes, S.M.; Lee, H.; Heestand, G.M.; et al. Single-Molecule Methylation Profiles of Cell-Free DNA in Cancer with Nanopore Sequencing. Genome Med. 2023, 15, 33.
- Cescon, D.W.; Bratman, S.V.; Chan, S.M.; Siu, L.L. Circulating Tumor DNA and Liquid Biopsy in Oncology. Nat. Cancer 2020, 1, 276–290.
- Igari, F.; Tanaka, H.; Giuliano, A.E. The Applications of Plasma Cell-Free DNA in Cancer Detection: Implications in the Management of Breast Cancer Patients. Crit. Rev. Oncol. Hematol. 2022, 175, 103725.
- Smith, J.T.; Balar, A.; Lakhani, D.A.; Kluwe, C.; Zhao, Z.; Kopparapu, P.; Almodovar, K.; Muterspaugh, A.; Yan, Y.; York, S.; et al. Circulating Tumor DNA as a Biomarker of Radiographic Tumor Burden in SCLC. JTO Clin. Res. Rep. 2021, 2, 100110.
- Mohan, S.; Foy, V.; Ayub, M.; Leong, H.S.; Schofield, P.; Sahoo, S.; Descamps, T.; Kilerci, B.; Smith, N.K.; Carter, M.; et al. Profiling of Circulating Free DNA Using Targeted and Genome-Wide Sequencing in Patients with SCLC. J. Thorac. Oncol. 2020, 15, 216–230.
- Almodovar, K.; Iams, W.T.; Meador, C.B.; Zhao, Z.; York, S.; Horn, L.; Yan, Y.; Hernandez, J.; Chen, H.; Shyr, Y.; et al. Longitudinal Cell-Free DNA Analysis in Patients with Small Cell Lung Cancer Reveals Dynamic Insights into Treatment Efficacy and Disease Relapse. J. Thorac. Oncol. 2018, 13, 112–123.
- Tolmeijer, S.H.; Boerrigter, E.; Sumiyoshi, T.; Kwan, E.M.; Ng, S.W.S.; Annala, M.; Donnellan, G.; Herberts, C.; Benoist, G.E.; Hamberg, P.; et al. Early On-Treatment Changes in Circulating Tumor DNA Fraction and Response to Enzalutamide or Abiraterone in Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2023, 29, 2835–2844.
- Semenkovich, N.P.; Szymanski, J.J.; Earland, N.; Chauhan, P.S.; Pellini, B.; Chaudhuri, A.A. Genomic Approaches to Cancer and Minimal Residual Disease Detection Using Circulating Tumor DNA. J. Immunother. Cancer 2023, 11, e006284.
- Gatto, L.; Franceschi, E.; Di Nunno, V.; Tosoni, A.; Lodi, R.; Brandes, A.A. Liquid Biopsy in Glioblastoma Management: From Current Research to Future Perspectives. Oncologist 2021, 26, 865–878.
- Mair, R.; Mouliere, F. Cell-Free DNA Technologies for the Analysis of Brain Cancer. Br. J. Cancer 2022, 126, 371–378.
- Liu, L.; Toung, J.M.; Jassowicz, A.F.; Vijayaraghavan, R.; Kang, H.; Zhang, R.; Kruglyak, K.M.; Huang, H.J.; Hinoue, T.; Shen, H.; et al. Targeted Methylation Sequencing of Plasma Cell-Free DNA for Cancer Detection and Classification. Ann. Oncol. 2018, 29, 1445–1453.
- Sadeh, R.; Sharkia, I.; Fialkoff, G.; Rahat, A.; Gutin, J.; Chappleboim, A.; Nitzan, M.; Fox-Fisher, I.; Neiman, D.; Meler, G.; et al. ChIP-Seq of Plasma Cell-Free Nucleosomes Identifies Gene Expression Programs of the Cells of Origin. Nat. Biotechnol. 2021, 39, 586–598.
- Baca, S.C.; Seo, J.-H.; Davidsohn, M.P.; Fortunato, B.; Semaan, K.; Sotudian, S.; Lakshminarayanan, G.; Diossy, M.; Qiu, X.; El Zarif, T.; et al. Liquid Biopsy Epigenomic Profiling for Cancer Subtyping. Nat. Med. 2023, 29, 2737–2741.
- Nassiri, F.; Chakravarthy, A.; Feng, S.; Shen, S.Y.; Nejad, R.; Zuccato, J.A.; Voisin, M.R.; Patil, V.; Horbinski, C.; Aldape, K.; et al. Detection and Discrimination of Intracranial Tumors Using Plasma Cell-Free DNA Methylomes. Nat. Med. 2020, 26, 1044–1047.
- Burgener, J.M.; Zou, J.; Zhao, Z.; Zheng, Y.; Shen, S.Y.; Huang, S.H.; Keshavarzi, S.; Xu, W.; Liu, F.-F.; Liu, G.; et al. Tumor-Naïve Multimodal Profiling of Circulating Tumor DNA in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2021, 27, 4230–4244.
- Stackpole, M.L.; Zeng, W.; Li, S.; Liu, C.-C.; Zhou, Y.; He, S.; Yeh, A.; Wang, Z.; Sun, F.; Li, Q.; et al. Cost-Effective Methylome Sequencing of Cell-Free DNA for Accurately Detecting and Locating Cancer. Nat. Commun. 2022, 13, 5566.
- Galardi, F.; De Luca, F.; Romagnoli, D.; Biagioni, C.; Moretti, E.; Biganzoli, L.; Di Leo, A.; Migliaccio, I.; Malorni, L.; Benelli, M. Cell-Free DNA-Methylation-Based Methods and Applications in Oncology. Biomolecules 2020, 10, 1677.
- Fu, S.; Debes, J.D.; Boonstra, A. DNA Methylation Markers in the Detection of Hepatocellular Carcinoma. Eur. J. Cancer 2023, 191, 112960.
- Luo, H.; Wei, W.; Ye, Z.; Zheng, J.; Xu, R. Liquid Biopsy of Methylation Biomarkers in Cell-Free DNA. Trends Mol. Med. 2021, 27, 482–500.
- Shen, S.Y.; Burgener, J.M.; Bratman, S.V.; De Carvalho, D.D. Preparation of CfMeDIP-Seq Libraries for Methylome Profiling of Plasma Cell-Free DNA. Nat. Protoc. 2019, 14, 2749–2780.
- deVos, T.; Tetzner, R.; Model, F.; Weiss, G.; Schuster, M.; Distler, J.; Steiger, K.V.; Grutzmann, R.; Pilarsky, C.; Habermann, J.K.; et al. Circulating Methylated SEPT9 DNA in Plasma Is a Biomarker for Colorectal Cancer. Clin. Chem. 2009, 55, 1337–1346.
- Müller, D.; Győrffy, B. DNA Methylation-Based Diagnostic, Prognostic, and Predictive Biomarkers in Colorectal Cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2022, 1877, 188722.
- Weiss, G.; Schlegel, A.; Kottwitz, D.; König, T.; Tetzner, R. Validation of the SHOX2/PTGER4 DNA Methylation Marker Panel for Plasma-Based Discrimination between Patients with Malignant and Nonmalignant Lung Disease. J. Thorac. Oncol. 2017, 12, 77–84.
- Oh, T.; Kim, N.; Moon, Y.; Kim, M.S.; Hoehn, B.D.; Park, C.H.; Kim, T.S.; Kim, N.K.; Chung, H.C.; An, S. Genome-Wide Identification and Validation of a Novel Methylation Biomarker, SDC2, for Blood-Based Detection of Colorectal Cancer. J. Mol. Diagn. 2013, 15, 498–507.
- Klein, E.A.; Madhavan, S.; Beer, T.M.; Bettegowda, C.; Liu, M.C.; Hartman, A.-R.; Hackshaw, A. Dying to Find Out: The Cost of Time at the Dawn of the Multicancer Early Detection Era. Cancer Epidemiol. Biomark. Prev. 2023, 32, 1003–1010.
- Park, B.H.; Shen, S.Y.; Min, J.; Fleshner, N.; Knox, J.; May, T.; Ailles, L.; Newton, Y.; Zhang, J.; Singhania, R. Development of a Genome-Wide Methylome Enrichment Platform for Multi-Cancer Early Detection (MCED). Cancer Res. 2023, 83 (Suppl. S7), 1030.
- Huang, M.; He, J.; Lai, W.; Liu, L.; Xu, H.; Zeng, Y.; Lan, Q.; Lin, X.; Chu, Z. Methylated Septin 9 Gene Is an Important Prognostic Marker in Stage II and Stage III Colorectal Cancer for Evaluating Local Recurrence or Distant Metastasis after Surgery. BMC Gastroenterol. 2022, 22, 87.
- Hier, J.; Vachon, O.; Bernstein, A.; Ibrahim, I.; Mlynarek, A.; Hier, M.; Alaoui-Jamali, M.A.; Maschietto, M.; da Silva, S.D. Portrait of DNA Methylated Genes Predictive of Poor Prognosis in Head and Neck Cancer and the Implication for Targeted Therapy. Sci. Rep. 2021, 11, 10012.
- Ko, K.; Kananazawa, Y.; Yamada, T.; Kakinuma, D.; Matsuno, K.; Ando, F.; Kuriyama, S.; Matsuda, A.; Yoshida, H. Methylation Status and Long-Fragment Cell-Free DNA Are Prognostic Biomarkers for Gastric Cancer. Cancer Med. 2021, 10, 2003–2012.
- Bae, J.M.; Shin, S.-H.; Kwon, H.-J.; Park, S.-Y.; Kook, M.C.; Kim, Y.-W.; Cho, N.-Y.; Kim, N.; Kim, T.-Y.; Kim, D.; et al. ALU and LINE-1 Hypomethylations in Multistep Gastric Carcinogenesis and Their Prognostic Implications. Int. J. Cancer 2012, 131, 1323–1331.
- Ul Haq, S.; Schmid, S.; Aparnathi, M.K.; Hueniken, K.; Zhan, L.J.; Sacdalan, D.; Li, J.J.N.; Meti, N.; Patel, D.; Cheng, D.; et al. Cell-Free DNA Methylation-Defined Prognostic Subgroups in Small-Cell Lung Cancer Identified by Leukocyte Methylation Subtraction. iScience 2022, 25, 105487.
- Rini, B.I.; Zhang, J.; Hall, O.; Bergener, J.; Wang, Y.; Brown, B.; Min, J.; Shen, S.Y.; Fleshner, N.; Polio, A.; et al. 1910P Evaluation of a Genome-Wide Methylome Enrichment Platform for Circulating Tumor DNA Quantification and Prognostic Performance in Renal Cell Carcinoma (RCC). Ann. Oncol. 2023, 34, S1028.
- Liu, G.; Zhang, J.; Hall, O.; Bergener, J.; Wang, Y.; Brown, B.; Min, J.; Shen, S.Y.; Pienkowski, M.; Huang, S.H.; et al. 866P Prognostic Performance of a Genome-Wide Methylome Enrichment Platform in Head and Neck Cancer. Ann. Oncol. 2023, 34, S561.
- Majchrzak-Celińska, A.; Paluszczak, J.; Kleszcz, R.; Magiera, M.; Barciszewska, A.-M.; Nowak, S.; Baer-Dubowska, W. Detection of MGMT, RASSF1A, P15INK4B, and P14ARF Promoter Methylation in Circulating Tumor-Derived DNA of Central Nervous System Cancer Patients. J. Appl. Genet. 2013, 54, 335–344.
- Lok, B.H.; Gardner, E.E.; Schneeberger, V.E.; Ni, A.; Desmeules, P.; Rekhtman, N.; de Stanchina, E.; Teicher, B.A.; Riaz, N.; Powell, S.N.; et al. PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 523–535.
- He, T.; Zhang, M.; Zheng, R.; Zheng, S.; Linghu, E.; Herman, J.G.; Guo, M. Methylation of SLFN11 Is a Marker of Poor Prognosis and Cisplatin Resistance in Colorectal Cancer. Epigenomics 2017, 9, 849–862.
- Stewart, C.A.; Tong, P.; Cardnell, R.J.; Sen, T.; Li, L.; Gay, C.M.; Masrorpour, F.; Fan, Y.; Bara, R.O.; Feng, Y. Dynamic Variations in Epithelial-to-Mesenchymal Transition (EMT), ATM, and SLFN11 Govern Response to PARP Inhibitors and Cisplatin in Small Cell Lung Cancer. Oncotarget 2017, 8, 28575.
- Murai, J.; Feng, Y.; Guoying, K.Y.; Ru, Y.; Tang, S.-W.; Shen, Y.; Pommier, Y. Resistance to PARP Inhibitors by SLFN11 Inactivation Can Be Overcome by ATR Inhibition. Oncotarget 2016, 7, 76534.
- Tserpeli, V.; Stergiopoulou, D.; Londra, D.; Giannopoulou, L.; Buderath, P.; Balgkouranidou, I.; Xenidis, N.; Grech, C.; Obermayr, E.; Zeillinger, R. Prognostic Significance of SLFN11 Methylation in Plasma Cell-Free DNA in Advanced High-Grade Serous Ovarian Cancer. Cancers 2021, 14, 4.
- André, T.; Cohen, R.; Salem, M.E. Immune Checkpoint Blockade Therapy in Patients with Colorectal Cancer Harboring Microsatellite Instability/Mismatch Repair Deficiency in 2022. In American Society of Clinical Oncology Educational Book; American Society of Clinical Oncology: Alexandria, VA, USA, 2022; Volume 42, pp. 233–241.
- Pasanen, A.; Loukovaara, M.; Bützow, R. Clinicopathological Significance of Deficient DNA Mismatch Repair and MLH1 Promoter Methylation in Endometrioid Endometrial Carcinoma. Mod. Pathol. 2020, 33, 1443–1452.
- Wang, D.; O’Rourke, D.; Sanchez-Garcia, J.F.; Cai, T.; Scheuenpflug, J.; Feng, Z. Development of a Liquid Biopsy Based Purely Quantitative Digital Droplet PCR Assay for Detection of MLH1 Promoter Methylation in Colorectal Cancer Patients. BMC Cancer 2021, 21, 797.
- Lu, Y.-T.; Plets, M.; Morrison, G.; Cunha, A.T.; Cen, S.Y.; Rhie, S.K.; Siegmund, K.D.; Daneshmand, S.; Quinn, D.I.; Meeks, J.J.; et al. Cell-Free DNA Methylation as a Predictive Biomarker of Response to Neoadjuvant Chemotherapy for Patients with Muscle-Invasive Bladder Cancer in SWOG S1314. Eur. Urol. Oncol. 2023, 6, 516–524.
- Luskin, M.R.; Murakami, M.A.; Manalis, S.R.; Weinstock, D.M. Targeting Minimal Residual Disease: A Path to Cure? Nat. Rev. Cancer 2018, 18, 255–263.
- Norton, L. Cancer Log-Kill Revisited. In American Society of Clinical Oncology Educational Book; American Society of Clinical Oncology: Alexandria, VA, USA, 2014; Volume 34, pp. 3–7.
- Blackburn, E.H. Cancer Interception. Cancer Prev. Res. 2011, 4, 787–792.
- Chen, X.; Zhang, J.; Ruan, W.; Huang, M.; Wang, C.; Wang, H.; Jiang, Z.; Wang, S.; Liu, Z.; Liu, C.; et al. Urine DNA Methylation Assay Enables Early Detection and Recurrence Monitoring for Bladder Cancer. J. Clin. Investig. 2020, 130, 6278–6289.
- van Zogchel, L.M.J.; Lak, N.S.M.; Verhagen, O.J.H.M.; Tissoudali, A.; Gussmalla Nuru, M.; Gelineau, N.U.; Zappeij-Kannengieter, L.; Javadi, A.; Zijtregtop, E.A.M.; Merks, J.H.M.; et al. Novel Circulating Hypermethylated RASSF1A DdPCR for Liquid Biopsies in Patients with Pediatric Solid Tumors. JCO Precis. Oncol. 2021, 5, 1738–1748.
- Yuan, Z.; Wang, S.; Ni, K.; Zhan, Y.; Ma, H.; Liu, X.; Xin, R.; Zhou, X.; Liu, Z.; Zhao, X. Circulating Methylated SEPT9 DNA Analyses to Predict Recurrence Risk and Adjuvant Chemotherapy Benefit in Stage II to III Colorectal Cancer. Med. Sci. Monit. 2022, 28, e937757-1.
- Mo, S.; Ye, L.; Wang, D.; Han, L.; Zhou, S.; Wang, H.; Dai, W.; Wang, Y.; Luo, W.; Wang, R.; et al. Early Detection of Molecular Residual Disease and Risk Stratification for Stage I to III Colorectal Cancer via Circulating Tumor DNA Methylation. JAMA Oncol. 2023, 9, 770–778.
- Leon Arellano, M.; García-Arranz, M.; Guadalajara, H.; Olivera-Salazar, R.; Valdes-Sanchez, T.; García-Olmo, D. Analysis of Septin 9 Gene Hypermethylation as Follow-Up Biomarker of Colorectal Cancer Patients after Curative Surgery. Diagnostics 2022, 12, 993.