Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Adrián Povo-Retana.
Trabectedin (TRB) and Lurbinectedin (LUR) are alkaloid compounds originally isolated from Ecteinascidia turbinata with proven antitumoral activity. Both molecules are structural analogues that differ on the tetrahydroisoquinoline moiety of the C subunit in TRB, which is replaced by a tetrahydro-β-carboline in LUR.
- trabectedin
- lurbinectedin
- macrophages
- lymphocytes
- ecteinascidins
- combined therapies
1. Introduction
Oceans and seas constitute 71% of the Earth’s surface and account for 90% of biodiversity on our planet. Marine ecosystems are composed of complex communities of animals, plants, fungi, and microorganisms such as bacteria, protozoa, algae, and chromists [1]. Hence, the marine biosphere is rising as a fundamental potential source of bioactive molecules [2].
Thousands of marine natural products with biological therapeutic relevance are identified every year; in 2017, 1490 novel compounds were reported in 477 articles and 1544 were reported in 2018, which were vastly documented in 469 publications [3].
There is a growing interest within the pharmaceutical field for drug screening, discovery, and development in aquatic ecosystems due to the secondary metabolites that are generated by marine organisms.
2. Ascidians as a Source of Bioactive Molecules: Ecteinascidia turbinata
Ascidians are ancestral marine urochordates and tunicates that are considered filter-feeders [4]. This is the reason why they are considered pollution indicators and present unique characteristics in the animal kingdom; these organisms produce alternative proteins, such as specific oxidases and phytochelatins, and synthesize cellulose.
This enormous family is composed of more than 3000 different species whose reproduction is both sexual and asexual and, up-to-date, these organisms have directly been identified as a source of more than 1200 bioactive molecules [5].
Thus, ascidians have drawn the attention of the biomedical field due to their ability to synthesize secondary metabolites. Attending to the chemical nature and molecular structure of these biomolecules, there are three main groups: alkaloids, peptides, and polyketides [5,6,7][5][6][7].
Alkaloids are the most prominent family of compounds that exert antimicrobial [6] and anti-tumour activities [1,5,8][1][5][8]. Within this group it is relevant to mention saframycins, jorumycins, renieramycins, and ecteinascidins that share structural similarities within the bis-tetrahydroisoquinoline chemical moieties [9]. They inhibit essential kinases that regulate the cell cycle, such as protein kinase B (PKB) and cyclin-dependent kinases (CDKs), and alter the mitochondrial inner membrane potential [5]. Trabectedin and lurbinectedin, the anti-tumour compounds marketed by PharmaMar, belong to this category. Peptides from two to eighteen amino acids constitute a smaller subset (5% of bioactive molecules) distributed into linear peptides, cyclic peptides, and depsipeptides (peptides composed by ester and amide bonds). Finally, a third group has been identified, polyketides, which are complex molecules built from simple carboxylic acids and synthesized by polyketide synthetases [5,10][5][10].
This is a simplified overview as, not only are these molecules synthesized by ascidians, but their associated symbiont microorganisms have a pivotal role in the production of these defensive molecules that protect these marine creatures from their natural predators [5,7,11,12][5][7][11][12].
Ascidians show enhanced cellular plasticity within the Chordate phylum, and for this reason, are used as regenerative biology models [13].
Ecteinascidia turbinata is a tunicate that normally inhabits the Caribbean Sea, Gulf of Mexico, Bermuda, East Coast of Florida, and it has been seen in the Mediterranean Sea in the warmest periods [4] (Figure 1). This organism and a symbiont, Candidatus Endoecteinascidia frumetensis, are the natural sources of trabectedin [6[6][11],11], and it was the first ascidian compound with an anti-tumour activity to receive both EU (EMA) and US (FDA) approval [4].

Figure 1. Ecteinascidia turbinata, the ascidian source for trabectedin and lurbinectedin (Courtesy of PharmaMar S.A., Madrid, Spain).
3. Trabectedin and Lurbinectedin Molecular Structures
Ecteinascidins’ structures were first reported in 1987 by Rinehart et al., although the anti-tumour activity of these compounds was reported in 1969 from tunicate total extracts; however, due to the limited availability of the primary source, it took almost two decades to identify the molecular structure of the compounds that exerted the anti-tumour activity [14].
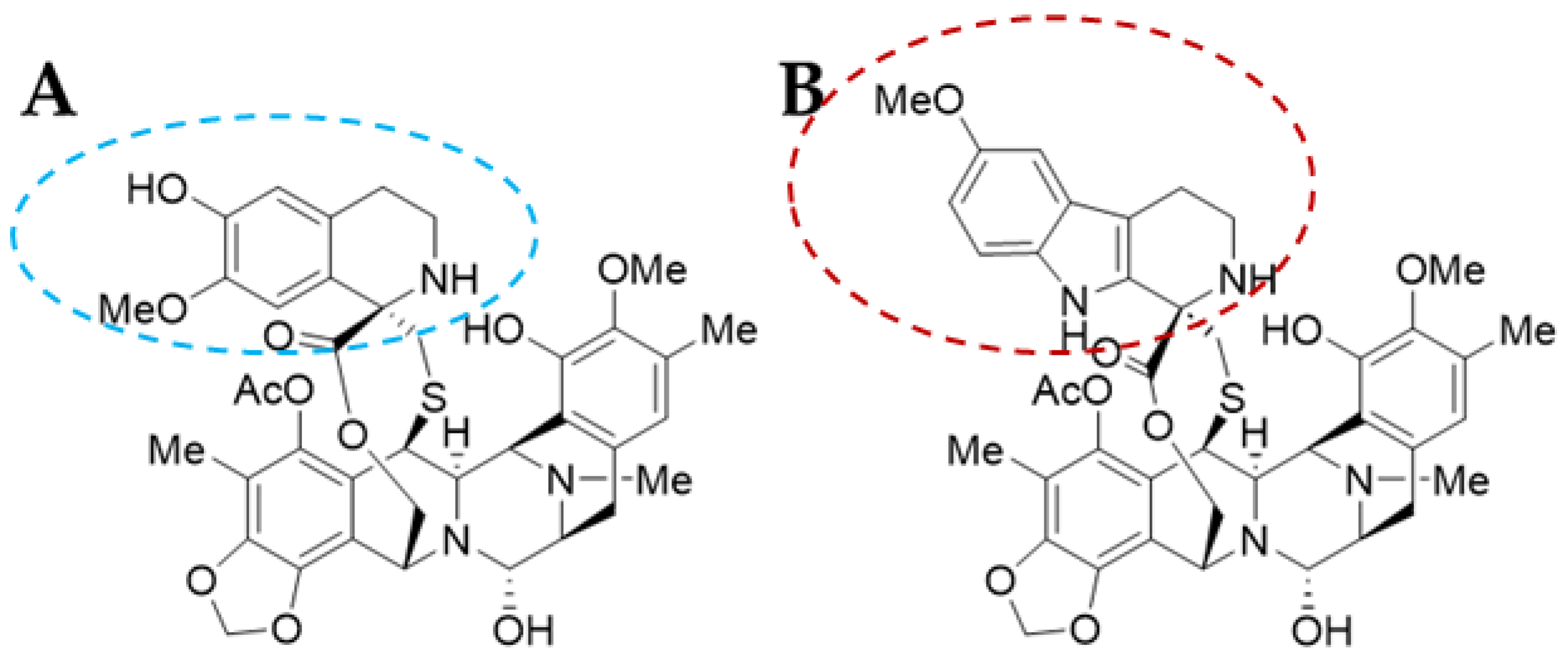
Trabectedin (ET-743 or TRB) was first isolated in 1990 and its X-ray crystallographic structure was resolved in 1992 [13]. It is a tetrahydroquinoline alkaloid with a molecular weight of 761.81 g/mol and a complex chemical structure composed of three fused tetrahydroisoquinoline rings (subunits A–C): a mono-bridged pentacyclic skeleton of two tetrahydroisoquinoline rings (subunits A and B) linked by a 10-member lactone bridge through a benzylic sulphide linkage attached to an additional ring by a -spyro ring to a third tetrahydroisoquinoline structure (Figure 2A). It is semi-synthetically produced and currently marketed as Yondelis® (Madrid, Spain) [15]. The complete synthesis of this compound by Ma and Chen’s group has recently been reviewed by Gao et al. [9], along with other tetrahydroisoquinoline alkaloid compounds.
Lurbinectedin (PM01183 or LUR) is a structural derivative of the former molecule. TRB and LUR differ in the C-subunit; TRB presents a tetrahydroisoquinoline (circled in blue) which is replaced by a tetrahydro-β-carboline (circled in red; Figure 2B) [16,17][16][17]. As a consequence, there are important pharmacodynamic and pharmacokinetic modifications [17]; LUR exhibits a distribution volume four times lower than TRB and exhibits a three-fold higher tolerance dose (MTD), presenting a distinct profile [18]. The LUR molecular structure is slightly larger. The molecular weight for PM01183 is 784.87 g/mol, and it is commercialized as Zepzelca® (Madrid, Spain).
4. Trabectedin and Lurbinectedin Uses in Oncology
TRB is indicated, in combination with pegylated liposomal doxorubicin, for patients with relapsed platinum-sensitive ovarian cancer [19,20[19][20][21],21], as well as for the treatment of advanced soft tissue sarcoma in adults [22,23,24,25][22][23][24][25] when ifosfamides and anthracyclines have failed [19,20][19][20].
TRB is applied for soft tissue sarcoma (STS) [26] and is applied in a phase III study of mesenchymal chondrosarcoma [27] as well as in a phase II study of extraskeletal myxoid chondrosarcoma. It is also used in non-operable liposarcomas and leiomyosarcomas [28,29,30,31,32,33,34][28][29][30][31][32][33][34] and it is applied for metastatic synovial sarcoma [35,36][35][36].
Recently, monocyte to lymphocyte ratio was demonstrated to be a prognostic tool in the treatment of STS with trabectedin. This ratio could be easily applied in clinical practice to assess TRB efficacy [37].
Lurbinectedin (LUR) was approved by the FDA in June 2020 after a phase II/III trial for the treatment of small cell lung carcinoma [38]. Additional assays were conducted for the treatment of ovarian cancer, breast cancer (ongoing in patients of BRCA1 loss of function), sarcoma, and acute myeloid leukaemia [39,40,41,42,43][39][40][41][42][43].
5. Mechanism of Action of Trabectedin and Lurbinectedin
Several mechanisms of action have been described for TRB and LUR. The most extensively documented mechanisms are related to their role as DNA-binding agents and transcriptional modulators. Still, there is mounting evidence suggesting that, apart from the well-known molecular mechanisms that are affected in tumoral cells (described below), the impact on the immune system compartment is of pivotal importance in the outcome of patients who are treated with these ecteinascidins [44,45][44][45].
5.1. TRB and LUR Act as DNA Intercalating Agents and Transcriptional Regulators
Both compounds bind to guanines in the DNA minor groove, specifically to the exocyclic N2 amino moiety through an in situ iminium intermediate that is formed by the dehydration of the carbinolamine that is located in the A subunit. Thus, TRB and LUR act as intercalating DNA agents [45]. The DNA-TRB or LUR adduct is additionally stabilised by existing van der Waals interactions and hydrogen bonds between the A and B subunits. This covalent, newly formed interaction induces a DNA torsion towards the major groove; this feature seems to be unique to these families of compounds [17,46,47][17][46][47].
Combinatorial chemical substitutions revealed that the carbinolamine moiety is relevant to the pharmacological activity of these molecules since derived compounds (ET-745) lacking this functional group fail to bind to DNA. Both anti-tumour drugs induce transcription-dependent stress and genomic instability [48]. In addition, this interaction with the DNA interferes with the transcription of genes whose promoter regions contain CG-rich sequences. Moreover, this transcriptional regulation is implemented by the dephosphorylation, ubiquitination, and degradation of the RNA polymerase II (Pol II) on the DNA template [49].
5.2. TRB and LUR Affect Homologous Recombination (HR) and Nucleotide Excision Repair (NER) DNA Repair Mechanisms
The main DNA repairing mechanism that is affected by TRB and LUR is nucleotide excision repair (NER). When there is a lack of this process, the cytotoxic capacity of these molecules over tumour cells is diminished [50]. If the affected mechanism is homologous recombination (HR), as it occurs in decreased expression of BRCA1/2, the reported cytotoxic effect is higher. TRB and LUR inhibit both NER and HR in tumoral cells [51]. Two of the principal components of the NER mechanism are XPG (Xeroderma pigmentosum group G) and XPF (Xeroderma pigmentosum group F) endonucleases that cleave the damaged DNA double-strand and correct the lesion [52]. In vivo studies in the yeast S. pombe showed that Rad13, a homolog protein, forms a tertiary complex, Rad13-DNA-TRB, that induces double-strand breaks, leading to the activation of programmed cell death processes [53].
5.3. TRB and LUR Affect Transcription, Cell Cycle, and Induce Apoptosis in Tumour Cell Lines
The steric hindrance induced by the tertiary complex prevents transcription factor binding to conserved consensus DNA sequences where there is an enrichment in GC. A dose-dependent binding inhibition has been reported at low micromolar doses (50–300 µM) for TBP, E2F, SRF, and CCAAT transcription factors by gel shift assays, and at even lower concentrations for NF-Y (10–30 µM). TRB induced a decrease of nucleosomes at 100 nM [54,55][54][55].
NF-Y is a central transcription factor that mediates the activation of the human gene that codes for P-glycoprotein or MDR-1, which recruits histone acetyltransferase PCAF to the MDR-1 promoter. TRB abrogates its transcriptional activation [56,57][56][57] and in doing so, it prevents ABCB1 channel activity, and therefore, it avoids the multidrug resistance that is associated with the overexpression of this protein in tumour cells [58,59,60][58][59][60].
In the low nanomolar range, TRB inhibits B2 cyclin transcription which might explain G2 cell cycle blockade [55,61][55][61]. It activates non-dependent P53 apoptosis and produces a cell cycle blockade in the late S and G2-M phases [53,61][53][61].
These anti-tumour compounds trigger RNA polymerase dephosphorylation and facilitate RNA polymerase II degradation via ubiquitination [16[16][62][63][64],62,63,64], drastically modulating messenger RNA transcription [16,65][16][65].
TRB and LUR induce apoptosis by both the intrinsic and extrinsic pathways in lung cancer A549 cell lines [66], and in MCF-7 and MDA-MB-453 breast cancer cells [67] in a time- and concentration-dependent fashion. It has been proposed that lurbinectedin in monotherapy is more effective for relapsed SCLC than other approved therapies [68,69,70][68][69][70].
5.4. TRB and LUR Regulate Tumour Microenvironment
It has extensively been reported that these anti-tumour drugs impact the tumour microenvironment (TME), target human tumour-associated macrophages (hTAM) [46,71[46][71][72][73],72,73], and inhibit the transcription of pro-inflammatory cytokines such as CCL2 (chemokine ligand 2), IL-6 [74], VEGF [65], CCL3, CCL7, and CCL14 [53,75][53][75]. TRB and LUR are known to modulate the immune response within the tumour microenvironment by specifically targeting mononuclear phagocytes [44,65,73,76,77][44][65][73][76][77]. Furthermore, it has recently been shown that TRB and LUR modulate the macrophage electrophysiology and polarisation state towards a proinflammatory-like (M1) activation state in quiescent macrophages, suggesting that TAMs pro-inflammatory re-education occurs in murine peritoneal rodent macrophages [78]. Moreover, LUR effectively eliminates both cancer cells and cancer stem cells in preclinical models of uterine cervical cancer [79]. Human TAMs are functionally inhibited and depleted by TRB, which improves the anti-tumour adaptative response to anti-PD-1 therapy [80].
5.5. TRB and LUR Affect the Human Immune System
Both TRB and LUR exert a direct impact on all the immune cell subsets, which probably contributes to the therapeutic actions of these drugs. Nevertheless, adverse effects have occasionally been observed in oncological patients, constituting an exclusion criterion for patients undergoing treatment with these anti-tumour drugs [75,81,82,83][75][81][82][83]. At this point, the development of prognostic biomarkers associated with the appearance of adverse effects after treatment with these drugs is a relevant area of research. Additionally, both drugs have been proposed to be applied in combination with immune checkpoint inhibitors, along with a plethora of specific targeted therapies, as addressed in Section 7.
5.5.1. Impact on Phagocytes/Myeloid Compartment (Neutrophils, Monocyte/Macrophages, DCs)
The most common adverse effect of TRB or LUR administration is neutropenia, which is reported in one-third of cancer patients undergoing these treatments, and if it is severe, it constitutes a motive for withdrawal. Both drugs target the mononuclear phagocytic system, specifically, monocytes and macrophages. They can inhibit cytoskeleton dynamics and motility, phagocytosis, and efferocytosis, and trigger apoptosis, as well as the recruitment of monocytes to the tumour site and induce apoptosis [65,84,85,86][65][84][85][86].
There are no currently available studies on dendritic cells regarding TRB and LUR. It would be very interesting to evaluate the dual role of these cells in tumour immunity [87,88,89,90,91,92][87][88][89][90][91][92]. However, TRB antitumoral activity was assessed in an orthotopic xenograft murine model bearing a doxorubicin-resistant follicular dendritic cell sarcoma derived from a patient and it was concluded that this tumour was slightly sensitive to TRB, but it was not statistically significant [93].
It has been proposed, extensively studied, and reviewed that these anti-tumour molecules exert “tropism” for hTAM [44]. They inhibit angiogenesis by inhibiting the expression of VEGF, PDGF, FGF, and metastasis by regulating MMPs, and abolish the immunosuppression that is established within the TME. In this sense, LUR has been identified as an inhibitor of myeloid suppressor cells, both in vivo and in vitro [79,94][79][94].
The impact of TRB and LUR on human macrophages has been extensively reported: these antitumoral compounds induce programmed cell death in sensitive macrophages [86] and, in those that retain viability, it favours a pro-inflammatory-like activated state. These compounds upregulate HLA (MHC class I and II) transcripts, glycolysis, NF-κB, and P38 proinflammatory pathways, and induce mitochondrial biogenesis. Additionally, both antitumoral drugs activate PPP and increase NADPH-oxidase-dependent ROS production as well as O2− generation and induce a rupture of the TCA cycle at MDH and IDH, favouring the metabolic HIF-1α stabilisation. This metabolic reconfiguration leads to the canonical hMφ proinflammatory activation [95]. TNFα and IL-8 are augmented in the supernatant of primary hMφ [96] (Figure 3).
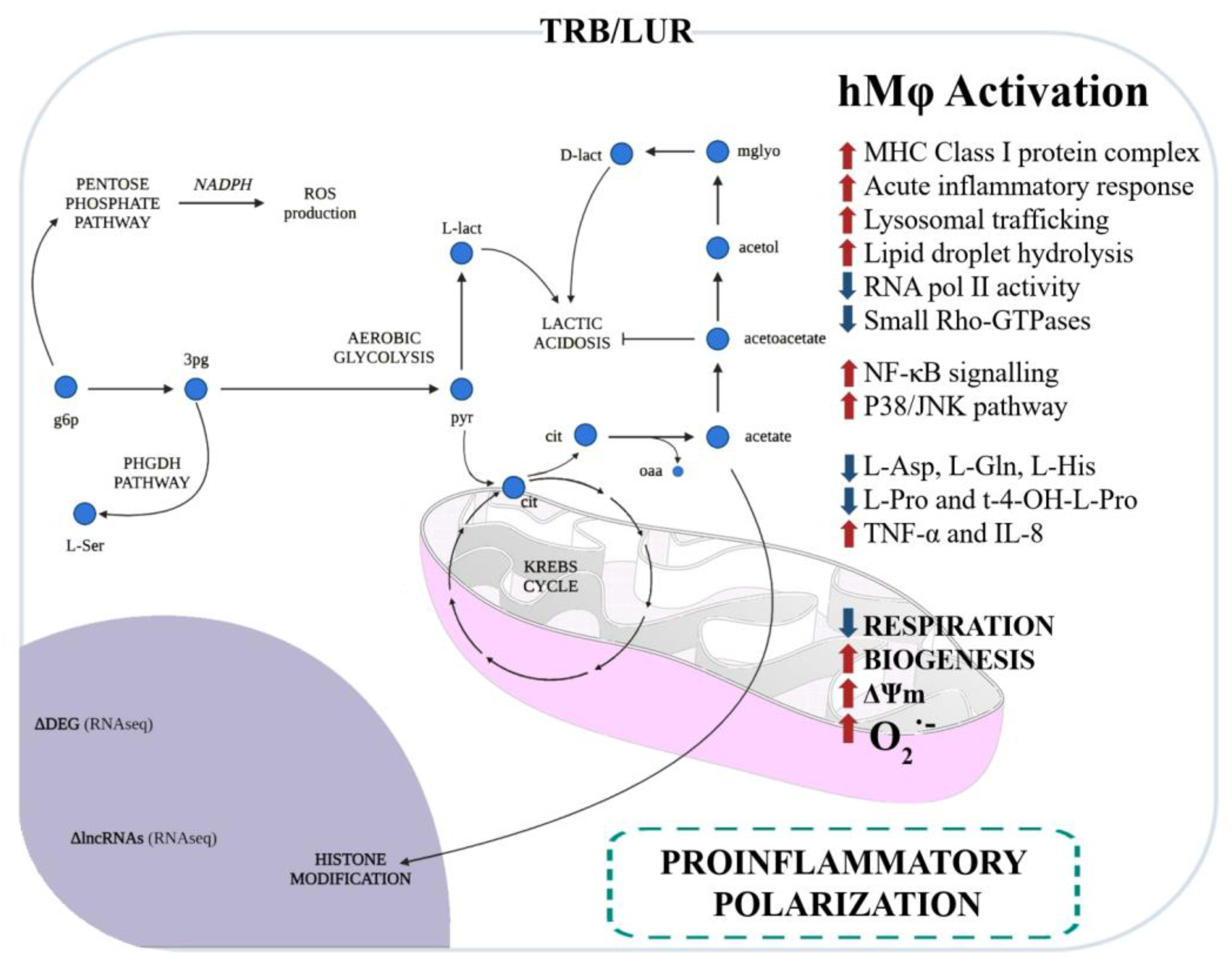
Figure 3. Immunometabolic and functional response of human macrophages to trabectedin and lurbinectedin. Glycolysis and the pentose phosphate pathway (PPP) are favoured, and serine production is predicted by RNAseq and fluxomic approaches [96].
Hence, not only do these ecteinascidins impact the tumour microenvironment and the tumour itself, but they also trigger a proinflammatory activation, at least in in vitro primary human macrophages’ cell culture. Macrophage polarisation is driven by macrophage metabolism, and it regulates the biological function of these innate cells [95,97,98][95][97][98]. The understanding of these processes is crucial to comprehend the interplay between immune cells and the tumour and its microenvironment to design specific targeted therapies to improve oncologic patient outcomes and overall survival, as well as progression-free survival.
5.5.2. Impact on Lymphoid Subsets (T Cells, B Cells, NK Cells, and NKT Cells)
These molecules activate NK cells; TRB triggers direct and NK-mediated cytotoxicity in multiple myeloma [99], and both TRB and LUR exert a cytotoxic effect targeting B cells in Chronic Lymphocytic Leukaemia (CLL) [43,100][43][100]. TRB exhibited cytotoxic effects in diffuse large B cell leukaemia [101]. TRB and LUR have been shown to activate CD4+ and CD8+ T-cells as well, promoting the adaptive anti-tumour immune response, inducing their infiltration in vivo and the proliferation of activated effector T-cells in vitro [43,100,102,103][43][100][102][103].
Globally, TRB and LUR functionally/mechanistically exert three major roles: these drugs induce apoptosis in tumour cells and stromal supporting cells (TAMs), modulate the TME, and instruct both innate and adaptive immune cells towards an anti-tumour-activated phenotype. Thus, they directly kill tumour cells and prevent the immunosuppressive milieu that is established. These molecules potentiate the anti-tumour immune response to neutralise the tumour.
It has been suggested that the expression of TRAIL-R in the different leukocyte subsets is related to the mechanism of TRB-induced apoptosis and could be useful to explain the differential viability effects on cell viability of each of the myeloid and lymphoid subsets [44,100][44][100].
Figure 4 recapitulates the most extensively reported effects of TRB and LUR on tumour cells, the tumour microenvironment, and tumour-supporting cells, as well as immune cell activation, and is supported by experimental evidence [86,96][86][96].
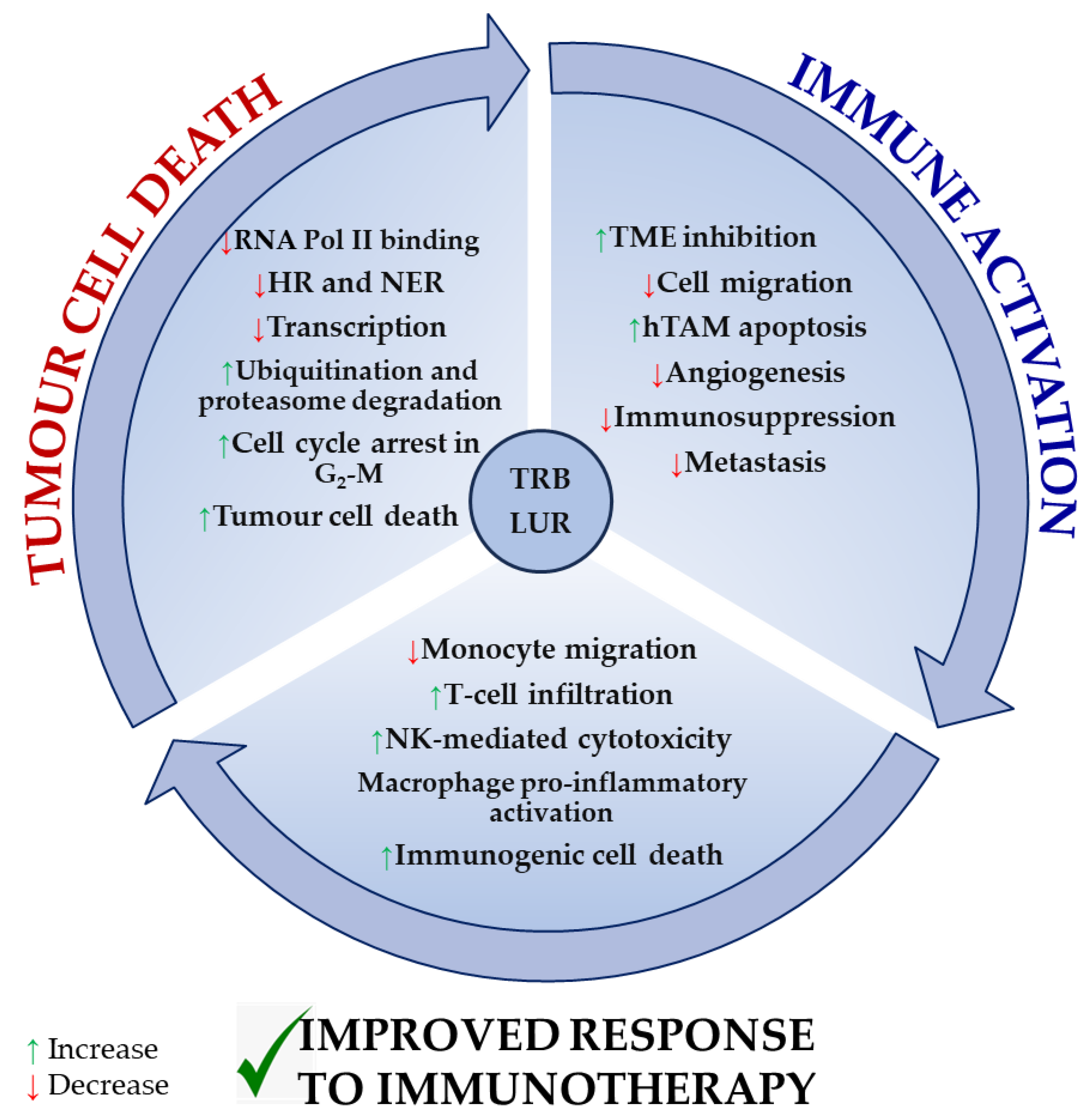
Figure 4. Reported molecular mechanisms for TRB and LUR in tumour cells, the tumour microenvironment, and immune cells. TRB and LUR are DNA intercalating molecules and transcriptional regulators. They impact human TAM biology acting as TME regulators and immunomodulate human immune response and activation.
Both drugs exert a direct cytotoxic effect in tumour cells by interfering with the transcription machinery and cell cycle and inducing immunogenic tumour cell death. As a result, they inhibit the immunosuppressive milieu that is normally established by the tumour and tumour supporting cells. TRB and LUR downregulate the expression of VEGF and several metalloproteases, preventing both tumour progression and metastasis and simultaneously activating NK-mediated cytotoxicity, T-cell infiltration (in vivo), and macrophage proinflammatory activation (in vitro). They reduce monocyte migration (Figure 4).
Taken together, these data strongly suggest that TRB and LUR elicit a higher immune response through two different paths: these drugs prevent the functional pro-tumoral immune suppression in the TME and favour immune cell activation, which explains tumour regression and overall patient improvement and makes these molecules great candidates for their combination with immunotherapy. Therefore, it is highly relevant to establish and explain how these ecteinascidins modulate the human adaptative immune response since mounting evidence demonstrates that, not only do these drugs induce tumour cell death, but they also instruct the immune system to activate and respond to neutralise the tumour.
6. Novel Functional and Molecular Targets for Trabectedin
Trabectedin induces ferroptosis via HIF-1α/IRP1/TFR1 and Keap1/Nrf2/GPX4 in non-small cell lung cancer cells (nSCLC) [104]. This biological process is proven to be essential in macrophage function and is arising as a novel target in oncology that is increasingly becoming a hot topic within the molecular oncology and immunotherapy field. NRF2 and redox biology seem to be regulated by the molecular mechanism of TRB [96[96][104],104], although this observation deserves further investigation.
It has additionally been nominated as a potential candidate for drug repositioning in the FDA for type II diabetes treatment by docking. It has been postulated as an α-glucosidase inhibitor with an in vitro IC50 of 1.2 ± 0.7 µM, alongside demeclocycline. Nonetheless, this repositioning needs to be further assessed due to its systemic toxicity, hence, a well-justified safety study ought to be conducted [105].
Recently, it has been shown that TRB inhibited therapy induced senescence in tumours by altering glutamine metabolism [106].
7. Combination Therapies Involving Trabectedin and Lurbinectedin
There is a pressing need to design combination treatments that may include conventional chemotherapeutic agents, immunologically targeted therapies such as immune checkpoint inhibitors (ICIs), or specific inhibitor molecules that target signaling or metabolic regulators, due to the complexity of tumoral biology and adaptation capacity, as well as resistance generation. In this sense, the field is experiencing a remarkable expansion: globally, TRB is assessed in combination with ICIs (antiPD-1/PD-L1 [107,108,109,110][107][108][109][110] and/or CTLA-4 [109]), monoclonal antibodies (-mAbs) that may act as either molecular inhibitors or activators, specific inhibitors of molecular targets (PARP [51[51][111][112][113],111,112,113], MDM2 [114], VEGF [115,116[115][116][117],117], CCR5 [118], m-TOR [119], IGF1-R [120], BCL2 [121], ATM/ATR [122],PPAR-γ [123], and CK-2/CLK2 [124]), recombinant proteins (shTRAIL [125]), topoisomerase inhibitors (irinotecan [126,127,128[126][127][128][129][130][131],129,130,131], topotecan [127], and camptothecin [132]), and immuno-modulatory biomolecules such as L19-mTNF [133] or dexamethasone [134], combined with propranolol [135], a β-adrenergic receptor inhibitor, or Wnt/β-catenin inhibitors [136] (PRI-724). It is combined with physical agents (hyperthermia [137] and radiation [138[138][139][140][141],139,140,141], among other strategies) as it is shown in Table 1, where it is indicated in which pathologies and cellular or murine models are applied.
Table 1.
Combination therapies involving trabectedin.
| Category | Treatment | Co-Treatment Function | Type of Cancer | Ref. |
|---|---|---|---|---|
| Monoclonal antibodies (mAb) | TRB + bevacizumab | Anti–VEGF | Partially platinum-sensitive recurrent ovarian cancer | [115] |
| TRB + AVE1642 | Anti–IGF1R | Ewing sarcoma | [120] | |
| TRB + VE-821 + KU-60019 | Anti–ATR (VE-821) Anti–ATM (KU-60019) |
Cervical carcinoma, ovarian carcinoma | [122 | |
| TRB + ipilimumab + nivolumab | ||||
| Anti–CTLA-4 (ipilimumab) | ||||
| Anti–PD-1 (nivolumab) | Advanced soft tissue sarcoma | [109] |
109]. TRB + irinotecan has proven to be effective on a desmoplastic small round cell tumour patient-derived xenograft [130[130][131],131], cisplatin-resistant osteosarcoma [129], and rhabdomyosarcoma [128]. TRB + β-blocker propranolol combination has proven to be effective in vitro and ex vivo evaluations in cervical cancer in patient-derived organoids [135].
In the ovarian cancer cell model, TRB + anti-PD-1-mab showed synergistic efficacy [102], favouring the activation of effector CD4+ and CD8+ T-cells in vivo by the upregulation of IFN-γ and inducing a decrease of immunosuppressive MDSCs and regulatory T-cells [103,142][103][142]. Three dose levels of TRB + durvalumab (PD-L1 inhibitor) showed promising efficacy in a phase Ib multicentre trial (TRAMUNE) in relapsed platinum-refractory ovarian cancer [107,166][107][166].
A similar approach was conducted in murine osteosarcoma models where TRB inhibited osteosarcoma primary tumour growth and metastasis and enhanced the number of T-cell tumour-infiltrated cells (both CD4+ and CD8+). TRB induced the overexpression of PD-1 in vivo but it did not in vitro, and Chiara Ratti et al. proved that TRB + anti-PD-1 blocking antibody increased CD8+ infiltrating cells and TRB efficacy, whereas anti-PD-1 alone did not reduce osteosarcoma growth. The combination further increased CD4+ and CD8+ recruitment, shifted CD4+ naïve T cells to CD4+ effector memory cells, and rendered a higher efficacy compared to TRB alone, preventing osteosarcoma progression. This combination enhanced the expression of CTLA-4, suggesting that it might be a third suitable partner for combined immunotherapy [167].
TRB + everolimus was synergistic in cisplatin and paclitaxel-resistant ovarian clear cell carcinoma cell lines and mice xenografts [119]. TRB + maraviroc (CCR5 antagonist) was effective in classical Hodgkin lymphoma mesenchymal stromal cells [118]. In a phase II clinical trial, TRB + dexamethasone improved safety in pre-treated soft tissue sarcoma patients [134]. TRB + mitotane reduced invasiveness and metastatic processes in adrenocortical carcinoma [145]. TRB + metformin + CB2 emerged as a novel venue for diabetes-associated breast cancer in cell lines and xenograft murine models [144]. TRB + PRI-724 [136] and TRB + RG7112 [114] were effective in human in vitro soft tissue sarcoma cell lines (in MDM2-amplified liposarcoma and fibrosarcoma cell lines [114] and STS cell lines and primary cultures [136]). TRB + radiotherapy combination has been assayed in A549 and HT-29 cell lines [138]. This approach has been identified as beneficial for STS patients, especially when tumour sinkage for symptomatic relief is required [139], and for phase I and phase II clinical trials for patients with myxoid liposarcoma. In the first one, there is an improvement in both safety and antitumoral activity [141]; in the second one, the primary endpoint was not achieved but the combination was well-tolerated and effective in terms of pathological response.
LUR has been combined with several agents as it is shown in Table 2 where, again, it is indicated in which pathologies/cellular or murine models the combinations are applied. LUR has been combined with irinotecan in ovarian cell clear carcinoma cell lines showing synergistic effects [154] and in a case report showing BRCA-mutated platinum-resistant ovarian cancer patients had exceptional clinical responses [153]. LUR + olaparib (PARP inhibitor), in a phase I clinical trial for advanced solid tumours, is feasible and exhibited a disease control rate of 72.6% [157]. LUR + doxorubicin improved safety in a phase III clinical trial of SCLC [164], and in a phase II clinical trial, a benefit was observed in several types of metastatic and unresectable sarcomas [41]. In an expanded phase I clinical trial for advanced endometrial cancer, this combination favoured a better response rate as its duration progressed [163].
Table 2.
Combination therapies involving lurbinectedin.
| Category | Treatment | Co-Treatment Function | Type of Cancer | Ref. | |||
|---|---|---|---|---|---|---|---|
| Monoclonal antibodies (mAb) | LUR + VE-821 + KU-60019 | Anti–ATR (VE-821) Anti–ATM (KU-60019) |
Cervical carcinoma, ovarian carcinoma | [122] | |||
| Immune checkpoint inhibitors (ICIs) | LUR + αPD-1 + αCTLA-4 | Osteosarcoma, fibrosarcoma, lung cancer, breast cancer | [152] | ||||
| Inhibitors | ] | ||||||
| LUR + irinotecan | Topoisomerase I inhibitor | Ovarian clear cell carcinoma | [ | 154] | Immune checkpoint inhibitors (ICIs) | TRB + durvalumab | Anti–PD-L1 |
| Platinum-refractory ovarian carcinoma | [ | 107 | ] | ||||
| BRCA-mutated platinum-resistant ovarian cancer patient | [ | 153] | TRB + avelumab | Anti–PD-L1 | Advanced liposarcoma and leiomyosarcoma | [110] | |
| LUR + olaparib | PARP inhibitor | Advanced solid tumours | [157] | TRB + nivolumab + talimogene laherparepvec (TVEC) | |||
| LUR + berzosertib | Anti–PD-1 (nivolumab) Replication within tumours and production of GM-CSF (TVEC) |
Advanced previously treated sarcomas | ATR inhibitor[108] | ||||
| Small-cell lung cancer | [ | 155 | ] | TRB + α-PD-1 mAb | |||
| Biological agents | LUR + doxorubicin | Treatment of several sarcomas | Relapsed small-cell lung cancer | [164] | Ovarian cancer | [ | |
| Leiomyosarcoma, dedifferentiated liposarcoma, myxoid liposarcoma, synovial sarcoma, and desmoplastic small round cell tumour | 142 | ] | |||||
| [ | 41 | ] | Inhibitors | TRB + olaparib | PARP inhibitor | Breast cancer | |
| Recurrent advanced endometrial cancer | [163] | [ | 111] | ||||
| Advanced and unresectable bone and soft-tissue sarcomas | [112] | ||||||
| LUR + capecitaine | Treatment of metastatic colorectal cancer (mCRC) and metastatic breast cancer (MBC) | Metastatic breast cancer | [162] | Ewing sarcoma | [113] | ||
| Osteosarcoma, leiomyosarcoma | [143] | ||||||
| TRB + RG7112 | MDM2 antagonist | Soft tissue sarcoma | [114] | ||||
| LUR + paclitaxel | Treatment of several sarcoma | Small cell lung cancer, breast cancer, endometrial cancer | [158] | TRB + rucaparib | |||
| LUR + paclitaxel + bevacizumab | Anti–VEGF (bevacizumab) | Epithelial ovarian cancer | [158] | PARP inhibitor | Soft tissue sarcoma, dedifferentiated liposarcoma |
[51] | |
| TRB + PRI-724 | Wnt/β-Catenin inhibitor | Soft tissue sarcoma | [136] | ||||
| TRB + ponatinib | Multi-tyrosine kinase inhibitor | Solitary fibrous tumour of the pleura | [116] | ||||
| TRB + propranolol | β-adrenergic receptors antagonist | ||||||
| LUR + cisplatin | Mesothelioma | [160] | |||||
| LUR + gemcitabine | Treatment of advanced pancreatic cancer | Advanced solid tumours | [161] | Cervical cancer, ovarian cancer | [135] | ||
| LUR + 4C9-DM1 | Antibody-drug conjugate (ADC) that targets c-Kit | Small cell lung cancer | [165]TRB + pioglitazone | PPARγ agonist | Myxoid liposarcoma | [123] | |
| TRB + topotecan | Topoisomerase I inhibitor | Ovarian clear cell carcinoma | [127] | ||||
| TRB + irinotecan | Topoisomerase I inhibitor | Ovarian clear cell carcinoma | [127] | ||||
| Rhabdomyosarcoma | [128] | ||||||
| Cisplatin-resistant osteosarcoma | [129] | ||||||
| Relapsed desmoplastic small round cell tumour | [131] | ||||||
| Desmoplastic small round cell tumour | [130] | ||||||
| TRB + everolimus | mTOR inhibition | Cisplatin-resistant and paclitaxel-resistant ovarian clear cell carcinoma | [119] | ||||
| TRB + maraviroc | CCR5 antagonist | Classical Hodgkin lymphoma-mesenchymal stromal cells | [118] | ||||
| TRB + metformin + CB-2 |
Hypoglycemic agent (metformin) MCT4 inhibitor (CB-2) |
Diabetes-associated breast cancer | [144] | ||||
| TRB + camptothecin | Topoisomerase I inhibitor | Myxoid/round cell liposarcoma, undifferentiated pleomorphic sarcoma | [132] | ||||
| TRB + obatoclax | Bcl-2 inhibitor | Malignant pleural mesothelioma | [121] | ||||
| TRB + ABT-199 | Bcl-2 inhibitor | Malignant pleural mesothelioma | [121] | ||||
| TRB + OSI-906 | IGF1R inhibitor | Ewing sarcoma | [120] | ||||
| TRB + silmitasertib | CK2/CLK double-inhibitor | Uveal melanoma | [124] | ||||
| TRB + cabozantinib | c-MET/TAM (TYRO3, Axl, MERTK) receptor inhibitor | Uveal melanoma | [124] | ||||
| Biological agents | TRB + FOLFIRI (leucovorin + 5-fluorouracil + irinotecan) | Treatment of colorectal cancer | Colorectal cancer | [126] | |||
| TRB + mitotane | Treatment of adrenocortical carcinoma | Adrenocortical carcinoma | [145] | ||||
| TRB + dexamethasone | Glucocorticoid medication | Advanced/metastatic soft tissue sarcoma | [134] | ||||
| TRB + gemcitabine | Treatment of advanced pancreatic cancer can disrupt DNA replication and activate the S phase checkpoint | Pancreatic cancer | [146] | ||||
| TRB + paclitaxel | Treatment of advanced solid tumours | Advanced solid tumours | [147] | ||||
| TRB + docetaxel | Treatment of ovarian and peritoneal cancer | Recurrent/persistent ovarian and peritoneal cancer | [148] | ||||
| TRB + enterolactone | Anti-angiogenic activity | Epithelial ovarian cancer | [117] | ||||
| TRB + cisplatin | Treatment of malignant pleural mesothelioma | Malignant pleural mesothelioma | [121] | ||||
| TRB + carboplatin | Treatment of advanced solid tumours | Advanced solid tumours | [149] | ||||
| TRB + shTRAIL | Targets cancer cells to induce apoptosis | Colon cancer | [125] | ||||
| TRB + pAXL × CD3ε | Redirects T-lymphocyte cytotoxicity to AXL-expressing cells | Osteosarcoma | [150] | ||||
| TRB + L19-mTNF | Pro-inflammatory cytokine | Fibrosarcoma | [133] | ||||
| Physical agents | TRB + radiotherapy | Lung cancer, colon cancer | [138] | ||||
| Advanced soft tissue sarcoma | [139] | ||||||
| Localized resectable myxoid liposarcoma | [140,141][140][141] | ||||||
| Retroperitoneal leiomyosarcoma | [151] | ||||||
| TRB + hyperthermia | Osteosarcoma, liposarcoma, synovial sarcoma | [137] |
LUR has been evaluated in combination with ICIs (anti-PD-L1 and anti-CTLA-4 [152]) and in combination with irinotecan [153[153][154],154], ATR [122,155][122][155] alone or combined with ATM [156] and PARP [157] inhibitors, anti-VEGF [158] combined with cisplatin [83[83][159][160],159,160], paclitaxel [158], gemcitabine [161], capecitabine [162], doxorubicin [41[41][163][164],163,164], and immunomodulatory biomolecules such as antibody-drug complexes commonly referred to as ADCs (4C9-DM1 that targets c-Kit [165]).
TRB in combination with Anti-AXLxCD3ε has proven to be more effective than TRB alone in sarcoma cells [150]. TNT treatment (talimogene laherparepvec, nivolumab, and trabectedin) has shown to be synergistic against advanced sarcoma [108]. There is an ongoing phase I/II SAINT study using ipilimumab (CTLA-4 inhibitor), nivolumab (PD-1 inhibitor), and trabectedin, as a first-line treatment for advanced soft tissue sarcoma (ASTS) (NCT03138161) [
LUR + capecitabine was applied in a phase I trial for relapsed metastatic breast cancer (HR+) with promising results [162]. LUR + paclitaxel showed synergistic antitumoral activity and improved safety in a phase I trial in SCLC, breast and endometrial cancer patients, and in combination with bevacizumab (anti-VEGF) for epithelial ovarian cancer [158]. TRB or LUR + VE-821 + KU-60019 (anti-ATR and anti-ATM respectively) combinations were evaluated in ovarian and cervical cell lines and showed higher antitumoral activity, suggesting that this venue provided mechanistic evidence that could have potential therapeutic effects that need to be addressed [122]. For LUR in combination with ICIs: anti-PD-L1 and anti-CTLA-4 showed strong anti-neoplastic effects in osteosarcoma and fibrosarcoma cell lines, and breast cancer and fibrosarcoma murine models [152]. LUR + berzosertib (ATR inhibitor) showed synergy in SCLC in vivo, organoid, and in vitro models [155]. LUR + cisplatin showed promising activity in malignant pleural mesothelioma [160] but it was not feasible in advanced solid tumours due to toxicity issues [83]. TRB + gemcitabine was assessed in the phase I trial for several advanced tumour types showing well-tolerated results with higher antitumoral activity [161]. Finally, LUR + 4C9-DM1-ADC achieved a higher tumour growth inhibition rate compared to LUR alone in mice xenografts bearing human SCLC [165].
Interestingly, it has been proposed that these Ecteinascidins may be combined with specific antibodies forming antibody-drug complexes (ADC) [165] and nanoparticles [168]. These approaches might help to overcome unwanted collateral side effects and improve safety parameters, both locally in the vasculature at the injection site, and systemically.
References
- Mauro, M.; Lazzara, V.; Punginelli, D.; Arizza, V.; Vazzana, M. Antitumoral Compounds from Vertebrate Sister Group: A Review of Mediterranean Ascidians. Dev. Comp. Immunol. 2020, 108, 103669.
- Dou, X.; Dong, B. Origins and Bioactivities of Natural Compounds Derived from Marine Ascidians and Their Symbionts. Mar. Drugs 2019, 17, 670.
- Casertano, M.; Menna, M.; Imperatore, C. The Ascidian-Derived Metabolites with Antimicrobial Properties. Antibiotics 2020, 9, 510.
- Watters, D.J. Ascidian Toxins with Potential for Drug Development. Mar. Drugs 2018, 16, 162.
- Conte, M.; Fontana, E.; Nebbioso, A.; Altucci, L. Marine-Derived Secondary Metabolites as Promising Epigenetic Bio-Compounds for Anticancer Therapy. Mar. Drugs 2020, 19, 15.
- Wang, E.; Sorolla, M.A.; Gopal Krishnan, P.D.; Sorolla, A. From Seabed to Bedside: A Review on Promising Marine Anticancer Compounds. Biomolecules 2020, 10, 248.
- Gao, Y.; Tu, N.; Liu, X.; Lu, K.; Chen, S.; Guo, J. Progress in the Total Synthesis of Antitumor Tetrahydroisoquinoline Alkaloids. Chem. Biodivers. 2023, 20, e202300172.
- Amoutzias, G.; Chaliotis, A.; Mossialos, D. Discovery Strategies of Bioactive Compounds Synthesized by Nonribosomal Peptide Synthetases and Type-I Polyketide Synthases Derived from Marine Microbiomes. Mar. Drugs 2016, 14, 80.
- Le, V.H.; Inai, M.; Williams, R.M.; Kan, T. Ecteinascidins. A Review of the Chemistry, Biology and Clinical Utility of Potent Tetrahydroisoquinoline Antitumor Antibiotics. Nat. Prod. Rep. 2015, 32, 328–347.
- Matos, A.; Antunes, A. Symbiotic Associations in Ascidians: Relevance for Functional Innovation and Bioactive Potential. Mar. Drugs 2021, 19, 370.
- Gordon, T.; Shenkar, N. Solitary Ascidians as Model Organisms in Regenerative Biology Studies. Results Probl. Cell Differ. 2018, 65, 321–336.
- Aune, G.J.; Furuta, T.; Pommier, Y. Ecteinascidin 743: A Novel Anticancer Drug with a Unique Mechanism of Action. Anticancer Drugs 2002, 13, 545–555.
- Sakai, R.; Rinehart, K.L.; Guan, Y.; Wang, A.H.J. Additional Antitumor Ecteinascidins from a Caribbean Tunicate: Crystal Structures and Activities in Vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 11456–11460.
- D’Incalci, M.; Galmarini, C.M. A Review of Trabectedin (ET-743): A Unique Mechanism of Action. Mol. Cancer Ther. 2010, 9, 2157–2163.
- Cuevas, C.; Francesch, A. Development of Yondelis (Trabectedin, ET-743). A Semisynthetic Process Solves the Supply Problem. Nat. Prod. Rep. 2009, 26, 322–337.
- Soares, D.G.; Machado, M.S.; Rocca, C.J.; Poindessous, V.; Ouaret, D.; Sarasin, A.; Galmarini, C.M.; Henriques, J.A.P.; Escargueil, A.E.; Larsen, A.K. Trabectedin and Its C Subunit Modified Analogue PM01183 Attenuate Nucleotide Excision Repair and Show Activity toward Platinum-Resistant Cells. Mol. Cancer Ther. 2011, 10, 1481–1489.
- De Sanctis, R.; Jacobs, F.; Benvenuti, C.; Gaudio, M.; Franceschini, R.; Tancredi, R.; Pedrazzoli, P.; Santoro, A.; Zambelli, A. From Seaside to Bedside: Current Evidence and Future Perspectives in the Treatment of Breast Cancer Using Marine Compounds. Front. Pharmacol. 2022, 13, 909566.
- Markham, A. Lurbinectedin: First Approval. Drugs 2020, 80, 1345–1353.
- Pignata, S.; Pisano, C.; Di Napoli, M.; Cecere, S.C.; Tambaro, R.; Attademo, L. Treatment of Recurrent Epithelial Ovarian Cancer. Cancer 2019, 125, 4609–4615.
- Monk, B.J.; Herzog, T.J.; Wang, G.; Triantos, S.; Maul, S.; Knoblauch, R.; McGowan, T.; Shalaby, W.S.W.; Coleman, R.L. A Phase 3 Randomized, Open-Label, Multicenter Trial for Safety and Efficacy of Combined Trabectedin and Pegylated Liposomal Doxorubicin Therapy for Recurrent Ovarian Cancer. Gynecol. Oncol. 2020, 156, 535–544.
- D’Incalci, M. Trabectedin Mechanism of Action: What’s New? Future Oncol. 2013, 9, 5–10.
- Sheng, J.Y.; Movva, S. Systemic Therapy for Advanced Soft Tissue Sarcoma. Surg. Clin. N. Am. 2016, 96, 1141–1156.
- Nakamura, T.; Sudo, A. The Role of Trabectedin in Soft Tissue Sarcoma. Front. Pharmacol. 2022, 13, 777872.
- Miwa, S.; Yamamoto, N.; Hayashi, K.; Takeuchi, A.; Igarashi, K.; Tsuchiya, H. Therapeutic Targets for Bone and Soft-Tissue Sarcomas. Int. J. Mol. Sci. 2019, 20, 170.
- Meyer, M.; Seetharam, M. First-Line Therapy for Metastatic Soft Tissue Sarcoma. Curr. Treat. Options Oncol. 2019, 20, 6.
- Andreeva-Gateva, P.; Chakar, S. The Place of Trabectedin in the Treatment of Soft Tissue Sarcoma: An Umbrella Review of the Level One Evidence. Expert Opin. Orphan Drugs 2019, 7, 105–115.
- Morioka, H.; Takahashi, S.; Araki, N.; Sugiura, H.; Ueda, T.; Takahashi, M.; Yonemoto, T.; Hiraga, H.; Hiruma, T.; Kunisada, T.; et al. Results of Sub-Analysis of a Phase 2 Study on Trabectedin Treatment for Extraskeletal Myxoid Chondrosarcoma and Mesenchymal Chondrosarcoma. BMC Cancer 2016, 16, 479.
- Rubio, M.J.; Lecumberri, M.J.; Varela, S.; Alarcón, J.; Ortega, M.E.; Gaba, L.; Espinós, J.; Calzas, J.; Barretina, P.; Ruiz, I.; et al. Efficacy and Safety of Trabectedin in Metastatic Uterine Leiomyosarcoma: A Retrospective Multicenter Study of the Spanish Ovarian Cancer Research Group (GEICO). Gynecol. Oncol. Rep. 2020, 33, 100594.
- Jones, R.L.; Maki, R.G.; Patel, S.R.; Wang, G.; McGowan, T.A.; Shalaby, W.S.; Knoblauch, R.E.; von Mehren, M.; Demetri, G.D. Safety and Efficacy of Trabectedin When Administered in the Inpatient versus Outpatient Setting: Clinical Considerations for Outpatient Administration of Trabectedin. Cancer 2019, 125, 4435–4441.
- Vincenzi, B.; Napolitano, A.; Comandone, A.; Sanfilippo, R.; Celant, S.; Olimpieri, P.P.; Di Segni, S.; Russo, P.; Casali, P.G. Trabectedin Use in Soft-Tissue Sarcoma Patients in a Real-World Setting: Data from an Italian National Drug-Access Registry. Int. J. Cancer 2023, 152, 761–768.
- Zhou, M.Y.; Bui, N.Q.; Charville, G.W.; Ganjoo, K.N.; Pan, M. Treatment of De-Differentiated Liposarcoma in the Era of Immunotherapy. Int. J. Mol. Sci. 2023, 24, 9571.
- Nassif, E.F.; Keung, E.Z.; Thirasastr, P.; Somaiah, N. Myxoid Liposarcomas: Systemic Treatment Options. Curr. Treat. Options Oncol. 2023, 24, 274–291.
- Thirasastr, P.; Lin, H.; Amini, B.; Wang, W.L.; Cloutier, J.M.; Nassif, E.F.; Keung, E.Z.; Roland, C.L.; Feig, B.; Araujo, D.; et al. Retrospective Evaluation of the Role of Gemcitabine-Docetaxel in Well-Differentiated and Dedifferentiated Liposarcoma. Cancer Med. 2023, 12, 4282–4293.
- Gutierrez-Sainz, L.; Martinez-Fdez, S.; Pedregosa-Barbas, J.; Peña, J.; Alameda, M.; Viñal, D.; Villamayor, J.; Martinez-Recio, S.; Perez-Wert, P.; Pertejo-Fernandez, A.; et al. Efficacy of Second and Third Lines of Treatment in Advanced Soft Tissue Sarcomas: A Real-World Study. Clin. Transl. Oncol. 2023, 25, 3519–3526.
- Patel, N.; Pokras, S.; Ferma, J.; Casey, V.; Manuguid, F.; Culver, K.; Bauer, S. Treatment Patterns and Outcomes in Patients with Metastatic Synovial Sarcoma in France, Germany, Italy, Spain and the UK. Futur. Oncol. 2023, 19, 1261–1275.
- Okazaki, M.; Katano, K.; Sugita, H.; Tokoro, T.; Gabata, R.; Takada, S.; Nakanuma, S.; Makino, I.; Yagi, S. Early Progression of a Pancreatic Metastasis of Synovial Sarcoma after Pancreatectomy. Surg. Case Rep. 2023, 9, 30.
- Fausti, V.; De Vita, A.; Vanni, S.; Ghini, V.; Gurrieri, L.; Riva, N.; Casadei, R.; Maraldi, M.; Ercolani, G.; Cavaliere, D.; et al. Systemic Inflammatory Indices in Second-Line Soft Tissue Sarcoma Patients: Focus on Lymphocyte/Monocyte Ratio and Trabectedin. Cancers 2023, 15, 1080.
- Farago, A.F.; Drapkin, B.J.; Lopez-Vilarino de Ramos, J.A.; Galmarini, C.M.; Núñez, R.; Kahatt, C.; Paz-Ares, L. ATLANTIS: A Phase III Study of Lurbinectedin/Doxorubicin versus Topotecan or Cyclophosphamide/Doxorubicin/Vincristine in Patients with Small-Cell Lung Cancer Who Have Failed One Prior Platinum-Containing Line. Future Oncol. 2019, 15, 231–239.
- Poveda, A.; Del Campo, J.M.; Ray-Coquard, I.; Alexandre, J.; Provansal, M.; Guerra Alía, E.M.; Casado, A.; Gonzalez-Martin, A.; Fernández, C.; Rodriguez, I.; et al. Phase II Randomized Study of PM01183 versus Topotecan in Patients with Platinum-Resistant/Refractory Advanced Ovarian Cancer. Ann. Oncol. 2017, 28, 1280–1287.
- Cruz, C.; Llop-Guevara, A.; Garber, J.E.; Arun, B.K.; Pérez Fidalgo, J.A.; Lluch, A.; Telli, M.L.; Fernández, C.; Kahatt, C.; Galmarini, C.M.; et al. Multicenter Phase II Study of Lurbinectedin in BRCA-Mutated and Unselected Metastatic Advanced Breast Cancer and Biomarker Assessment Substudy. J. Clin. Oncol. 2018, 36, 3134–3143.
- Cote, G.M.; Choy, E.; Chen, T.; Marino-Enriquez, A.; Morgan, J.; Merriam, P.; Thornton, K.; Wagner, A.J.; Nathenson, M.J.; Demetri, G.; et al. A Phase II Multi-Strata Study of Lurbinectedin as a Single Agent or in Combination with Conventional Chemotherapy in Metastatic and/or Unresectable Sarcomas. Eur. J. Cancer 2020, 126, 21–32.
- Benton, C.B.; Chien, K.S.; Tefferi, A.; Rodriguez, J.; Ravandi, F.; Daver, N.; Jabbour, E.; Jain, N.; Alvarado, Y.; Kwari, M.; et al. Safety and Tolerability of Lurbinectedin (PM01183) in Patients with Acute Myeloid Leukemia and Myelodysplastic Syndrome. Hematol. Oncol. 2019, 37, 96–102.
- Risnik, D.; Colado, A.; Podaza, E.; Almejún, M.B.; Elías, E.E.; Bezares, R.F.; Fernández-Grecco, H.; Seija, N.; Oppezzo, P.; Borge, M.; et al. Immunoregulatory Effects of Lurbinectedin in Chronic Lymphocytic Leukemia. Cancer Immunol. Immunother. 2020, 69, 813–824.
- Allavena, P.; Belgiovine, C.; Digifico, E.; Frapolli, R.; D’Incalci, M. Effects of the Anti-Tumor Agents Trabectedin and Lurbinectedin on Immune Cells of the Tumor Microenvironment. Front. Oncol. 2022, 12, 851790.
- Gadducci, A.; Cosio, S. Trabectedin and Lurbinectedin: Mechanisms of Action, Clinical Impact, and Future Perspectives in Uterine and Soft Tissue Sarcoma, Ovarian Carcinoma, and Endometrial Carcinoma. Front. Oncol. 2022, 12, 914342.
- D’Incalci, M.; Zambelli, A. Trabectedin for the Treatment of Breast Cancer. Expert Opin. Investig. Drugs 2016, 25, 105–115.
- Hurley, L.H.; Zewail-Foote, M. The Antitumor Agent Ecteinascidin 743: Characterization of Its Covalent DNA Adducts and Chemical Stability. Adv. Exp. Med. Biol. 2001, 500, 289–299.
- Tumini, E.; Herrera-Moyano, E.; San Martín-Alonso, M.; Barroso, S.; Galmarini, C.M.; Aguilera, A. The Antitumor Drugs Trabectedin and Lurbinectedin Induce Transcription-Dependent Replication Stress and Genome Instability. Mol. Cancer Res. 2019, 17, 773–782.
- Santamaría Nuñez, G.; Robles, C.M.G.; Giraudon, C.; Martínez-Leal, J.F.; Compe, E.; Coin, F.; Aviles, P.; Galmarini, C.M.; Egly, J.-M. Lurbinectedin Specifically Triggers the Degradation of Phosphorylated RNA Polymerase II and the Formation of DNA Breaks in Cancer Cells. Mol. Cancer Ther. 2016, 15, 2399–2412.
- Damia, G.; Silvestri, S.; Carrassa, L.; Filiberti, L.; Faircloth, G.T.; Liberi, G.; Foiani, M.; D’Incalci, M. Unique Pattern of ET-743 Activity in Different Cellular Systems with Defined Deficiencies in DNA-Repair Pathways. Int. J. Cancer 2001, 92, 583–588.
- Laroche, A.; Chaire, V.; Le Loarer, F.; Algéo, M.P.; Rey, C.; Tran, K.; Lucchesi, C.; Italiano, A. Activity of Trabectedin and the PARP Inhibitor Rucaparib in Soft-Tissue Sarcomas. J. Hematol. Oncol. 2017, 10, 84.
- Herrero, A.B.; Martín-Castellanos, C.; Marco, E.; Gago, F.; Moreno, S. Cross-Talk between Nucleotide Excision and Homologous Recombination DNA Repair Pathways in the Mechanism of Action of Antitumor Trabectedin. Cancer Res. 2006, 66, 8155–8162.
- Allavena, P.; Signorelli, M.; Chieppa, M.; Erba, E.; Bianchi, G.; Marchesi, F.; Olimpio, C.O.; Bonardi, C.; Garbi, A.; Lissoni, A.; et al. Anti-Inflammatory Properties of the Novel Antitumor Agent Yondelis (Trabectedin): Inhibition of Macrophage Differentiation and Cytokine Production. Cancer Res. 2005, 65, 2964–2971.
- Bonfanti, M.; La Valle, E.; Fernandez Sousa Faro, J.M.; Faircloth, G.; Caretti, G.; Mantovani, R.; D’Incalci, M. Effect of Ecteinascidin-743 on the Interaction between DNA Binding Proteins and DNA. Anticancer Drug Des. 1999, 14, 179–186.
- D’Incalci, M.; Brunelli, D.; Marangon, E.; Simone, M.; Tavecchio, M.; Gescher, A.; Mantovani, R. Modulation of Gene Transcription by Natural Products—A Viable Anticancer Strategy. Curr. Pharm. Des. 2007, 13, 2744–2750.
- Jin, S.; Gorfajn, B.; Faircloth, G.; Scotto, K.W. Ecteinascidin 743, a Transcription-Targeted Chemotherapeutic That Inhibits MDR1 Activation. Proc. Natl. Acad. Sci. USA 2000, 97, 6775–6779.
- Barthomeuf, C.; Bourguet-Kondracki, M.-L.; Kornprobst, J.-M. Marine Metabolites Overcoming or Circumventing Multidrug Resistance Mediated by ATP-Dependent Transporters: A New Hope for Patient with Tumors Resistant to Conventional Chemotherapy. Anticancer Agents Med. Chem. 2012, 8, 886–903.
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the Role of Efflux Pumps in Multidrug-Resistant Cancer. Nat. Rev. Cancer 2019, 18, 452–464.
- Kodan, A.; Futamata, R.; Kimura, Y.; Kioka, N.; Nakatsu, T.; Kato, H.; Ueda, K. ABCB1/MDR1/P-Gp Employs an ATP-Dependent Twist-and-Squeeze Mechanism to Export Hydrophobic Drugs. FEBS Lett. 2021, 595, 707–716.
- Bossennec, M.; Di Roio, A.; Caux, C.; Ménétrier-Caux, C. MDR1 in Immunity: Friend or Foe? Oncoimmunology 2018, 7, e1499388.
- Erba, E.; Cavallaro, E.; Damia, G.; Mantovani, R.; Di Silvio, A.; Di Francesco, A.M.; Riccardi, R.; Cuevas, C.; Faircloth, G.T.; D’Incalci, M. The Unique Biological Features of the Marine Product Yondelis (ET-743, Trabectedin) Are Shared by Its Analog ET-637, Which Lacks the C Ring. Oncol. Res. 2004, 14, 579–587.
- Larsen, A.K.; Galmarini, C.M.; D’Incalci, M. Unique Features of Trabectedin Mechanism of Action. Cancer Chemother. Pharmacol. 2016, 77, 663–671.
- Aune, G.J.; Takagi, K.; Sordet, O.; Guirouilh-Barbat, J.; Antony, S.; Bohr, V.A.; Pommier, Y. Von Hippel-Lindau-Coupled and Transcription-Coupled Nucleotide Excision Repair-Dependent Degradation of RNA Polymerase II in Response to Trabectedin. Clin. Cancer Res. 2008, 14, 6449–6455.
- Feuerhahn, S.; Giraudon, C.; Martínez-Díez, M.; Bueren-Calabuig, J.A.; Galmarini, C.M.; Gago, F.; Egly, J.-M. XPF-Dependent DNA Breaks and RNA Polymerase II Arrest Induced by Antitumor DNA Interstrand Crosslinking-Mimetic Alkaloids. Chem. Biol. 2011, 18, 988–999.
- Belgiovine, C.; Bello, E.; Liguori, M.; Craparotta, I.; Mannarino, L.; Paracchini, L.; Beltrame, L.; Marchini, S.; Galmarini, C.M.; Mantovani, A.; et al. Lurbinectedin Reduces Tumour-Associated Macrophages and the Inflammatory Tumour Microenvironment in Preclinical Models. Br. J. Cancer 2017, 117, 628–638.
- Martínez-Serra, J.; Maffiotte, E.; Martín, J.; Bex, T.; Navarro-Palou, M.; Ros, T.; Plazas, J.M.; Vögler, O.; Gutiérrez, A.; Amat, J.C.; et al. Yondelis® (ET-743, Trabectedin) Sensitizes Cancer Cell Lines to CD95-Mediated Cell Death: New Molecular Insight into the Mechanism of Action. Eur. J. Pharmacol. 2011, 658, 57–64.
- Atmaca, H.; Bozkurt, E.; Uzunoglu, S.; Uslu, R.; Karaca, B. A Diverse Induction of Apoptosis by Trabectedin in MCF-7 (HER2-/ER+) and MDA-MB-453 (HER2+/ER-) Breast Cancer Cells. Toxicol. Lett. 2013, 221, 128–136.
- Petty, W.J.; Paz-Ares, L. Emerging Strategies for the Treatment of Small Cell Lung Cancer: A Review. JAMA Oncol. 2023, 9, 419–429.
- Bhamidipati, D.; Subbiah, V. Lurbinectedin, a DNA Minor Groove Inhibitor for Neuroendocrine Neoplasms beyond Small Cell Lung Cancer. Oncoscience 2023, 10, 22–23.
- Fudio, S.; Pérez-Ramos, L.; Asín-Prieto, E.; Zeaiter, A.; Lubomirov, R. A Model-Based Head-to-Head Comparison of Single-Agent Lurbinectedin in the Pivotal ATLANTIS Study. Front. Oncol. 2023, 13, 1152371.
- Germano, G.; Mantovani, A.; Allavena, P. Targeting of the Innate Immunity/Inflammation as Complementary Anti-Tumor Therapies. Ann. Med. 2011, 43, 581–593.
- Céspedes, M.V.; Guillén, M.J.; López-Casas, P.P.; Sarno, F.; Gallardo, A.; Álamo, P.; Cuevas, C.; Hidalgo, M.; Galmarini, C.M.; Allavena, P.; et al. Lurbinectedin Induces Depletion of Tumor-Associated Macrophages, an Essential Component of Its in Vivo Synergism with Gemcitabine, in Pancreatic Adenocarcinoma Mouse Models. Dis. Model. Mech. 2016, 9, 1461–1471.
- Allavena, P.; Germano, G.; Belgiovine, C.; D’Incalci, M.; Mantovani, A. Trabectedin: A Drug from the Sea That Strikes Tumor-Associated Macrophages. Oncoimmunology 2013, 2, e24614.
- Colmegna, B.; Uboldi, S.; Frapolli, R.; Licandro, S.A.; Panini, N.; Galmarini, C.M.; Badri, N.; Spanswick, V.J.; Bingham, J.P.; Kiakos, K.; et al. Increased Sensitivity to Platinum Drugs of Cancer Cells with Acquired Resistance to Trabectedin. Br. J. Cancer 2015, 113, 1687–1693.
- Germano, G.; Frapolli, R.; Simone, M.; Tavecchio, M.; Erba, E.; Pesce, S.; Pasqualini, F.; Grosso, F.; Sanfilippo, R.; Casali, P.G.; et al. Antitumor and Anti-Inflammatory Effects of Trabectedin on Human Myxoid Liposarcoma Cells. Cancer Res. 2010, 70, 2235–2244.
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell 2013, 23, 249–262.
- Castelli, C.; Rivoltini, L.; Rodolfo, M.; Tazzari, M.; Belgiovine, C.; Allavena, P. Modulation of the Myeloid Compartment of the Immune System by Angiogenic- and Kinase Inhibitor-Targeted Anti-Cancer Therapies. Cancer Immunol. Immunother. 2015, 64, 83–89.
- Peraza, D.A.; Povo-Retana, A.; Mojena, M.; García-Redondo, A.B.; Avilés, P.; Boscá, L.; Valenzuela, C. Trabectedin Modulates Macrophage Polarization in the Tumor-Microenvironment. Role of KV1.3 and KV1.5 Channels. Biomed. Pharmacother. 2023, 161, 114548.
- Yokoi, E.; Mabuchi, S.; Shimura, K.; Komura, N.; Kozasa, K.; Kuroda, H.; Takahashi, R.; Sasano, T.; Kawano, M.; Matsumoto, Y.; et al. Lurbinectedin (PM01183), a Selective Inhibitor of Active Transcription, Effectively Eliminates Both Cancer Cells and Cancer Stem Cells in Preclinical Models of Uterine Cervical Cancer. Investig. New Drugs 2019, 37, 818–827.
- Belgiovine, C.; Frapolli, R.; Liguori, M.; Digifico, E.; Colombo, F.S.; Meroni, M.; Allavena, P.; D’Incalci, M. Inhibition of Tumor-Associated Macrophages by Trabectedin Improves the Antitumor Adaptive Immunity in Response to Anti-PD-1 Therapy. Eur. J. Immunol. 2021, 51, 2677–2686.
- Trigo, J.; Subbiah, V.; Besse, B.; Moreno, V.; López, R.; Sala, M.A.; Peters, S.; Ponce, S.; Fernández, C.; Alfaro, V.; et al. Lurbinectedin as Second-Line Treatment for Patients with Small-Cell Lung Cancer: A Single-Arm, Open-Label, Phase 2 Basket Trial. Lancet Oncol. 2020, 21, 645–654.
- Subbiah, V.; Paz-Ares, L.; Besse, B.; Moreno, V.; Peters, S.; Sala, M.A.; López-Vilariño, J.A.; Fernández, C.; Kahatt, C.; Alfaro, V.; et al. Antitumor Activity of Lurbinectedin in Second-Line Small Cell Lung Cancer Patients Who Are Candidates for Re-Challenge with the First-Line Treatment. Lung Cancer 2020, 150, 90–96.
- Metaxas, Y.; Kahatt, C.; Alfaro, V.; Fudio, S.; Zeaiter, A.; Plummer, R.; Sessa, C.; Von Moos, R.; Forster, M.; Stathis, A. A Phase I Trial of Lurbinectedin in Combination with Cisplatin in Patients with Advanced Solid Tumors. Investig. New Drugs 2022, 40, 91–98.
- Jones, J.D.; Sinder, B.P.; Paige, D.; Soki, F.N.; Koh, A.J.; Thiele, S.; Shiozawa, Y.; Hofbauer, L.C.; Daignault, S.; Roca, H.; et al. Trabectedin Reduces Skeletal Prostate Cancer Tumor Size in Association with Effects on M2 Macrophages and Efferocytosis. Neoplasia 2019, 21, 172–184.
- Sinder, B.P.; Zweifler, L.; Koh, A.J.; Michalski, M.N.; Hofbauer, L.C.; Aguirre, J.I.; Roca, H.; McCauley, L.K. Bone Mass Is Compromised by the Chemotherapeutic Trabectedin in Association With Effects on Osteoblasts and Macrophage Efferocytosis. J. Bone Miner. Res. 2017, 32, 2116–2127.
- Povo-Retana, A.; Mojena, M.; Stremtan, A.B.; Fernández-García, V.B.; Gómez-Sáez, A.; Nuevo-Tapioles, C.; Molina-Guijarro, J.M.; Avendaño-Ortiz, J.; Cuezva, J.M.; López-Collazo, E.; et al. Specific Effects of Trabectedin and Lurbinectedin on Human Macrophage Function and Fate—Novel Insights. Cancers 2020, 12, 3060.
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865.
- Gardner, A.; de Mingo Pulido, Á.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924.
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24.
- Marciscano, A.E.; Anandasabapathy, N. The Role of Dendritic Cells in Cancer and Anti-Tumor Immunity. Semin. Immunol. 2021, 52, 101481.
- Mitchell, D.; Chintala, S.; Dey, M. Plasmacytoid Dendritic Cell in Immunity and Cancer. J. Neuroimmunol. 2018, 322, 63–73.
- Fu, C.; Jiang, A. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front. Immunol. 2018, 9, 3059.
- Oshiro, H.; Tome, Y.; Kiyuna, T.; Miyake, K.; Kawaguchi, K.; Higuchi, T.; Miyake, M.; Zang, Z.; Razmjooei, S.; Barangi, M.; et al. Temozolomide Targets and Arrests a Doxorubicin-Resistant Follicular Dendritic-Cell Sarcoma Patient-Derived Orthotopic Xenograft Mouse Model. Tissue Cell 2019, 58, 17–23.
- Kuroda, H.; Mabuchi, S.; Kozasa, K.; Yokoi, E.; Matsumoto, Y.; Komura, N.; Kawano, M.; Hashimoto, K.; Sawada, K.; Kimura, T. PM01183 Inhibits Myeloid-Derived Suppressor Cells In Vitro and In Vivo. Immunotherapy 2017, 9, 805–817.
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462.
- Povo-Retana, A.; Fariñas, M.; Landauro-Vera, R.; Mojena, M.; Alvarez-Lucena, C.; Fernández-Moreno, M.A.; Castrillo, A.; de la Rosa Medina, J.V.; Sánchez-García, S.; Foguet, C.; et al. Immunometabolic Actions of Trabectedin and Lurbinectedin on Human Macrophages: Relevance for Their Anti-Tumor Activity. Front. Immunol. 2023, 14, 1211068.
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism Governs Dendritic Cell and Macrophage Function. J. Exp. Med. 2016, 213, 15–23.
- Povo-Retana, A.; Landauro-Vera, R.; Fariñas, M.; Sánchez-García, S.; Alvarez-Lucena, C.; Marin, S.; Cascante, M.; Boscá, L. Defining the Metabolic Signatures Associated with Human Macrophage Polarisation. Biochem. Soc. Trans. 2023, 51, 1429–1436.
- Cucè, M.; Gallo Cantafio, M.E.; Siciliano, M.A.; Riillo, C.; Caracciolo, D.; Scionti, F.; Staropoli, N.; Zuccalà, V.; Maltese, L.; Di Vito, A.; et al. Trabectedin Triggers Direct and NK-Mediated Cytotoxicity in Multiple Myeloma. J. Hematol. Oncol. 2019, 12, 32.
- Banerjee, P.; Zhang, R.; Ivan, C.; Galletti, G.; Clise-Dwyer, K.; Barbaglio, F.; Scarfò, L.; Aracil, M.; Klein, C.; Wierda, W.; et al. Trabectedin Reveals a Strategy of Immunomodulation in Chronic Lymphocytic Leukemia. Cancer Immunol. Res. 2019, 7, 2036–2051.
- Spriano, F.; Chung, E.Y.; Panini, N.; Cascione, L.; Rinaldi, A.; Erba, E.; Stathis, A.; D’Incalci, M.; Bertoni, F.; Gatta, R. Trabectedin Is a Novel Chemotherapy Agent for Diffuse Large B Cell Lymphoma. Br. J. Haematol. 2019, 184, 1022–1025.
- Bailly, C.; Thuru, X.; Quesnel, B. Survey and Summary: Combined Cytotoxic Chemotherapy and Immunotherapy of Cancer: Modern Times. NAR Cancer 2020, 2, zcaa002.
- Zhao, T.; Zhu, Y.; Morinibu, A.; Kobayashi, M.; Shinomiya, K.; Itasaka, S.; Yoshimura, M.; Guo, G.; Hiraoka, M.; Harada, H. HIF-1-Mediated Metabolic Reprogramming Reduces ROS Levels and Facilitates the Metastatic Colonization of Cancers in Lungs. Sci. Rep. 2014, 4, 3793.
- Cai, S.; Ding, Z.; Liu, X.; Zeng, J. Trabectedin Induces Ferroptosis via Regulation of HIF-1α/IRP1/TFR1 and Keap1/Nrf2/GPX4 Axis in Non-Small Cell Lung Cancer Cells. Chem. Biol. Interact. 2023, 369, 110262.
- Rashid, R.S.M.; Temurlu, S.; Abourajab, A.; Karsili, P.; Dinleyici, M.; Al-Khateeb, B.; Icil, H. Drug Repurposing of FDA Compounds against α-Glucosidase for the Treatment of Type 2 Diabetes: Insights from Molecular Docking and Molecular Dynamics Simulations. Pharmaceuticals 2023, 16, 555.
- Pacifico, F.; Mellone, S.; D’Incalci, M.; Stornaiuolo, M.; Leonardi, A.; Crescenzi, E. Trabectedin Suppresses Escape from Therapy-Induced Senescence in Tumor Cells by Interfering with Glutamine Metabolism. Biochem. Pharmacol. 2022, 202, 115159.
- Toulmonde, M.; Brahmi, M.; Giraud, A.; Chakiba, C.; Bessede, A.; Kind, M.; Toulza, E.; Pulido, M.; Albert, S.; Guégan, J.P.; et al. Trabectedin plus Durvalumab in Patients with Advanced Pretreated Soft Tissue Sarcoma and Ovarian Carcinoma (TRAMUNE): An Open-Label, Multicenter Phase Ib Study. Clin. Cancer Res. 2022, 28, 1765–1772.
- Chawla, S.P.; Tellez, W.A.; Chomoyan, H.; Valencia, C.; Ahari, A.; Omelchenko, N.; Makrievski, S.; Brigham, D.A.; Chua-Alcala, V.; Quon, D.; et al. Activity of TNT: A Phase 2 Study Using Talimogene Laherparepvec, Nivolumab and Trabectedin for Previously Treated Patients with Advanced Sarcomas (NCT# 03886311). Front. Oncol. 2023, 13, 1116937.
- Gordon, E.M.; Chawla, S.P.; Tellez, W.A.; Younesi, E.; Thomas, S.; Chua-Alcala, V.S.; Chomoyan, H.; Valencia, C.; Brigham, D.A.; Moradkhani, A.; et al. SAINT: A Phase I/Expanded Phase II Study Using Safe Amounts of Ipilimumab, Nivolumab and Trabectedin as First-Line Treatment of Advanced Soft Tissue Sarcoma. Cancers 2023, 15, 906.
- Wagner, M.J.; Zhang, Y.; Cranmer, L.D.; Loggers, E.T.; Black, G.; McDonnell, S.; Maxwell, S.; Johnson, R.; Moore, R.; De Viveiros, P.H.; et al. A Phase 1/2 Trial Combining Avelumab and Trabectedin for Advanced Liposarcoma and Leiomyosarcoma. Clin. Cancer Res. 2022, 28, 2306–2312.
- Ávila-Arroyo, S.; Nuñez, G.S.; García-Fernández, L.F.; Galmarini, C.M. Synergistic Effect of Trabectedin and Olaparib Combination Regimen in Breast Cancer Cell Lines. J. Breast Cancer 2015, 18, 329–338.
- Grignani, G.; D’Ambrosio, L.; Pignochino, Y.; Palmerini, E.; Zucchetti, M.; Boccone, P.; Aliberti, S.; Stacchiotti, S.; Bertulli, R.; Piana, R.; et al. Trabectedin and Olaparib in Patients with Advanced and Non-Resectable Bone and Soft-Tissue Sarcomas (TOMAS): An Open-Label, Phase 1b Study from the Italian Sarcoma Group. Lancet Oncol. 2018, 19, 1360–1371.
- Ordóñez, J.L.; Amaral, A.T.; Carcaboso, A.M.; Herrero-Martín, D.; Del Carmen García-Macías, M.; Sevillano, V.; Alonso, D.; Pascual-Pasto, G.; San-Segundo, L.; Vila-Ubach, M.; et al. The PARP Inhibitor Olaparib Enhances the Sensitivity of Ewing Sarcoma to Trabectedin. Oncotarget 2015, 6, 18875–18890.
- Obrador-Hevia, A.; Martinez-Font, E.; Felipe-Abrio, I.; Calabuig-Farinãs, S.; Serra-Sitjar, M.; López-Guerrero, J.A.; Ramos, R.; Alemany, R.; Martín-Broto, J. RG7112, a Small-Molecule Inhibitor of MDM2, Enhances Trabectedin Response in Soft Tissue Sarcomas. Cancer Investig. 2015, 33, 440–450.
- Colombo, N.; Zaccarelli, E.; Baldoni, A.; Frezzini, S.; Scambia, G.; Palluzzi, E.; Tognon, G.; Lissoni, A.A.; Rubino, D.; Ferrero, A.; et al. Multicenter, Randomised, Open-Label, Non-Comparative Phase 2 Trial on the Efficacy and Safety of the Combination of Bevacizumab and Trabectedin with or without Carboplatin in Women with Partially Platinum-Sensitive Recurrent Ovarian Cancer. Br. J. Cancer 2019, 121, 744–750.
- Ghanim, B.; Baier, D.; Pirker, C.; Müllauer, L.; Sinn, K.; Lang, G.; Hoetzenecker, K.; Berger, W. Trabectedin Is Active against Two Novel, Patient-Derived Solitary Fibrous Pleural Tumor Cell Lines and Synergizes with Ponatinib. Cancers 2022, 14, 5602.
- Zeng, Z.; Lin, C.; Zhang, M.C.; Kossinna, P.; Wang, P.; Cao, D.; Wang, J.; Xu, M.; Wang, X.; Li, Q.; et al. Enterolactone and Trabectedin Suppress Epithelial Ovarian Cancer Synergistically via Upregulating THBS1. Phyther. Res. 2023, 37, 4722–4739.
- Casagrande, N.; Borghese, C.; Aldinucci, D. In Classical Hodgkin Lymphoma the Combination of the CCR5 Antagonist Maraviroc with Trabectedin Synergizes, Enhances DNA Damage and Decreases Three-Dimensional Tumor-Stroma Heterospheroid Viability. Haematologica 2022, 107, 287–291.
- Mabuchi, S.; Hisamatsu, T.; Kawase, C.; Hayashi, M.; Sawada, K.; Mimura, K.; Takahashi, K.; Takahashi, T.; Kurachi, H.; Kimura, T. The Activity of Trabectedin as a Single Agent or in Combination with Everolimus for Clear Cell Carcinoma of the Ovary. Clin. Cancer Res. 2011, 17, 4462–4473.
- Amaral, A.T.; Garofalo, C.; Frapolli, R.; Manara, M.C.; Mancarella, C.; Uboldi, S.; Di Giandomenico, S.; Ordóñez, J.L.; Sevillano, V.; Malaguarnera, R.; et al. Trabectedin Efficacy in Ewing Sarcoma Is Greatly Increased by Combination with Anti-IGF Signaling Agents. Clin. Cancer Res. 2015, 21, 1373–1382.
- Hoda, M.A.; Pirker, C.; Dong, Y.; Schelch, K.; Heffeter, P.; Kryeziu, K.; Van Schoonhoven, S.; Klikovits, T.; Laszlo, V.; Rozsas, A.; et al. Trabectedin Is Active against Malignant Pleural Mesothelioma Cell and Xenograft Models and Synergizes with Chemotherapy and Bcl-2 Inhibition in Vitro. Mol. Cancer Ther. 2016, 15, 2357–2369.
- Lima, M.; Bouzid, H.; Soares, D.G.; Selle, F.; Morel, C.; Galmarini, C.M.; Henriques, J.A.; Larsen, A.K.; Escargueil, A.E. Dual Inhibition of ATR and ATM Potentiates the Activity of Trabectedin and Lurbinectedin by Perturbing the DNA Damage Response and Homologous Recombination Repair. Oncotarget 2016, 7, 25885–25901.
- Frapolli, R.; Bello, E.; Ponzo, M.; Craparotta, I.; Mannarino, L.; Ballabio, S.; Marchini, S.; Carrassa, L.; Ubezio, P.; Porcu, L.; et al. Combination of PPARg Agonist Pioglitazone and Trabectedin Induce Adipocyte Differentiation to Overcome Trabectedin Resistance in Myxoid Liposarcomas. Clin. Cancer Res. 2019, 25, 7565–7575.
- Glinkina, K.; Nemati, F.; Teunisse, A.F.A.S.; Gelmi, M.C.; Etienne, V.; Kuipers, M.J.; Alsafadi, S.; Jager, M.J.; Decaudin, D.; Jochemsen, A.G. Preclinical Evaluation of Trabectedin in Combination with Targeted Inhibitors for Treatment of Metastatic Uveal Melanoma. Investig. Ophthalmol. Vis. Sci. 2022, 63, 14.
- Wang, X.; Wang, L.; Liu, W.; Liu, X.; Jia, X.; Feng, X.; Li, F.; Zhu, R.; Yu, J.; Zhang, H.; et al. Dose-Related Immunomodulatory Effects of Recombinant TRAIL in the Tumor Immune Microenvironment. J. Exp. Clin. Cancer Res. 2023, 42, 216.
- Zhu, G.; Zhao, M.; Han, Q.; Tan, Y.; Sun, Y.; Bouvet, M.; Clary, B.; Singh, S.R.; Ye, J.; Hoffman, R.M. Combination of Trabectedin with Irinotecan, Leucovorin and 5-Fluorouracil Arrests Primary Colorectal Cancer in an Imageable Patient-Derived Orthotopic Xenograft Mouse Model. Anticancer Res. 2019, 39, 6463–6470.
- Kawano, M.; Mabuchi, S.; Kishimoto, T.; Hisamatsu, T.; Matsumoto, Y.; Sasano, T.; Takahashi, R.; Sawada, K.; Takahashi, K.; Takahashi, T.; et al. Combination Treatment with Trabectedin and Irinotecan or Topotecan Has Synergistic Effects against Ovarian Clear Cell Carcinoma Cells. Int. J. Gynecol. Cancer 2014, 24, 829–837.
- Riccardi, A.; Meco, D.; Ubezio, P.; Mazzarella, G.; Marabese, M.; Faircloth, G.T.; Jimeno, J.; D’Incalci, M.; Riccardi, R. Combination of Trabectedin and Irinotecan Is Highly Effective in a Human Rhabdomyosarcoma Xenograft. Anticancer Drugs 2005, 16, 811–815.
- Higuchi, T.; Miyake, K.; Oshiro, H.; Sugisawa, N.; Yamamoto, N.; Hayashi, K.; Kimura, H.; Miwa, S.; Igarashi, K.; Chawla, S.P.; et al. Trabectedin and Irinotecan Combination Regresses a Cisplatinum-Resistant Osteosarcoma in a Patient-Derived Orthotopic Xenograft Nude-Mouse Model. Biochem. Biophys. Res. Commun. 2019, 513, 326–331.
- Zuco, V.; Pasquali, S.; Tortoreto, M.; Percio, S.; Doldi, V.; Barisella, M.; Collini, P.; Dagrada, G.P.; Brich, S.; Gasparini, P.; et al. Effectiveness of Irinotecan plus Trabectedin on a Desmoplastic Small Round Cell Tumor Patient-Derived Xenograft. DMM Dis. Model. Mech. 2023, 16, dmm049649.
- Ferrari, A.; Chiaravalli, S.; Bergamaschi, L.; Nigro, O.; Livellara, V.; Sironi, G.; Gasparini, P.; Pasquali, S.; Zaffaroni, N.; Stacchiotti, S.; et al. Trabectedin-Irinotecan, a Potentially Promising Combination in Relapsed Desmoplastic Small Round Cell Tumor: Report of Two Cases. J. Chemother. 2023, 35, 163–167.
- Martinez-Cruzado, L.; Tornin, J.; Rodriguez, A.; Santos, L.; Allonca, E.; Fernandez-Garcia, M.T.; Astudillo, A.; Garcia-Pedrero, J.M.; Rodriguez, R. Trabectedin and Campthotecin Synergistically Eliminate Cancer Stem Cells in Cell-of-Origin Sarcoma Models. Neoplasia 2017, 19, 460–470.
- Corbellari, R.; Nadal, L.; Villa, A.; Neri, D.; De Luca, R. The Immunocytokine L19-TNF Eradicates Sarcomas in Combination with Chemotherapy Agents or with Immune Check-Point Inhibitors. Anticancer Drugs 2020, 31, 799–805.
- Paz-Ares, L.; López-Pousa, A.; Poveda, A.; Balañá, C.; Ciruelos, E.; Bellmunt, J.; Del Muro, J.G.; Provencio, M.; Casado, A.; Rivera-Herrero, F.; et al. Trabectedin in Pre-Treated Patients with Advanced or Metastatic Soft Tissue Sarcoma: A Phase II Study Evaluating Co-Treatment with Dexamethasone. Investig. New Drugs 2012, 30, 729–740.
- Di Fonte, R.; Strippoli, S.; Garofoli, M.; Cormio, G.; Serratì, S.; Loizzi, V.; Fasano, R.; Arezzo, F.; Volpicella, M.; Derakhshani, A.; et al. Cervical Cancer Benefits from Trabectedin Combination with the β-Blocker Propranolol: In Vitro and Ex Vivo Evaluations in Patient-Derived Organoids. Front. Cell Dev. Biol. 2023, 11, 1178316.
- Martinez-Font, E.; Pérez-Capó, M.; Ramos, R.; Felipe, I.; Garcías, C.; Luna, P.; Terrasa, J.; Martín-Broto, J.; Vögler, O.; Alemany, R.; et al. Impact of Wnt/β-Catenin Inhibition on Cell Proliferation through Cdc25a Downregulation in Soft Tissue Sarcomas. Cancers 2020, 12, 2556.
- Harnicek, D.; Kampmann, E.; Lauber, K.; Hennel, R.; Cardoso Martins, A.S.; Guo, Y.; Belka, C.; Mörtl, S.; Gallmeier, E.; Kanaar, R.; et al. Hyperthermia Adds to Trabectedin Effectiveness and Thermal Enhancement Is Associated with BRCA2 Degradation and Impairment of DNA Homologous Recombination Repair. Int. J. Cancer 2016, 139, 467–479.
- Manda, K.; Präkelt, T.; Schröder, T.; Kriesen, S.; Hildebrandt, G. Radiosensitizing Effects of Trabectedin on Human A549 Lung Cancer Cells and HT-29 Colon Cancer Cells. Investig. New Drugs 2020, 38, 967–976.
- Hindi, N.; García, I.C.; Sánchez-Camacho, A.; Gutierrez, A.; Peinado, J.; Rincón, I.; Benedetti, J.; Sancho, P.; Santos, P.; Sánchez-Bustos, P.; et al. Trabectedin plus Radiotherapy for Advanced Soft-Tissue Sarcoma: Experience in Forty Patients Treated at a Sarcoma Reference Center. Cancers 2020, 12, 3740, Erratum in Cancers 2021, 13, 1557.
- Sanfilippo, R.; Hindi, N.; Cruz Jurado, J.; Blay, J.-Y.; Lopez-Pousa, A.; Italiano, A.; Alvarez, R.; Gutierrez, A.; Rincón-Perez, I.; Sangalli, C.; et al. Effectiveness and Safety of Trabectedin and Radiotherapy for Patients With Myxoid Liposarcoma: A Nonrandomized Clinical Trial. JAMA Oncol. 2023, 9, 656–663.
- Gronchi, A.; Hindi, N.; Cruz, J.; Blay, J.Y.; Lopez-Pousa, A.; Italiano, A.; Alvarez, R.; Gutierrez, A.; Rincón, I.; Sangalli, C.; et al. Trabectedin and RAdiotherapy in Soft Tissue Sarcoma (TRASTS): Results of a Phase I Study in Myxoid Liposarcoma from Spanish (GEIS), Italian (ISG), French (FSG) Sarcoma Groups. EClinicalMedicine 2019, 9, 35–43.
- Guo, Z.; Wang, H.; Meng, F.; Li, J.; Zhang, S. Combined Trabectedin and Anti-PD1 Antibody Produces a Synergistic Antitumor Effect in a Murine Model of Ovarian Cancer. J. Transl. Med. 2015, 13, 247.
- Pignochino, Y.; Capozzi, F.; D’Ambrosio, L.; Dell’Aglio, C.; Basiricò, M.; Canta, M.; Lorenzato, A.; Vignolo Lutati, F.; Aliberti, S.; Palesandro, E.; et al. PARP1 Expression Drives the Synergistic Antitumor Activity of Trabectedin and PARP1 Inhibitors in Sarcoma Preclinical Models. Mol. Cancer 2017, 16, 86.
- Hao, Q.; Huang, Z.; Li, Q.; Liu, D.; Wang, P.; Wang, K.; Li, J.; Cao, W.; Deng, W.; Wu, K.; et al. A Novel Metabolic Reprogramming Strategy for the Treatment of Diabetes-Associated Breast Cancer. Adv. Sci. 2022, 9, 2102303.
- Abate, A.; Rossini, E.; Bonini, S.A.; Fragni, M.; Cosentini, D.; Tiberio, G.A.M.; Benetti, D.; Hantel, C.; Laganà, M.; Grisanti, S.; et al. Cytotoxic Effect of Trabectedin In Human Adrenocortical Carcinoma Cell Lines and Primary Cells. Cancers 2020, 12, 928.
- Miao, X.; Koch, G.; Ait-Oudhia, S.; Straubinger, R.M.; Jusko, W.J. Pharmacodynamic Modeling of Cell Cycle Effects for Gemcitabine and Trabectedin Combinations in Pancreatic Cancer Cells. Front. Pharmacol. 2016, 7, 421.
- Chu, Q.; Mita, A.; Forouzesh, B.; Tolcher, A.W.; Schwartz, G.; Nieto, A.; Soto-Matos, A.; Alfaro, V.; Lebedinsky, C.; Rowinsky, E.K. Phase I and Pharmacokinetic Study of Sequential Paclitaxel and Trabectedin Every 2 Weeks in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2010, 16, 2656–2665.
- Monk, B.J.; Sill, M.W.; Hanjani, P.; Edwards, R.; Rotmensch, J.; De Geest, K.; Bonebrake, A.J.; Walker, J.L. Docetaxel plus Trabectedin Appears Active in Recurrent or Persistent Ovarian and Primary Peritoneal Cancer after up to Three Prior Regimens: A Phase II Study of the Gynecologic Oncology Group. Gynecol. Oncol. 2011, 120, 459–463.
- Vidal, L.; Magem, M.; Barlow, C.; Pardo, B.; Florez, A.; Montes, A.; Garcia, M.; Judson, I.; Lebedinsky, C.; Kaye, S.B.; et al. Phase i Clinical and Pharmacokinetic Study of Trabectedin and Carboplatin in Patients with Advanced Solid Tumors. Investig. New Drugs 2012, 30, 616–628.
- Polerà, N.; Mancuso, A.; Riillo, C.; Caracciolo, D.; Signorelli, S.; Grillone, K.; Ascrizzi, S.; Hokanson, C.A.; Conforti, F.; Staropoli, N.; et al. The First-In-Class Anti-AXL×CD3ε PronectinTM-Based Bispecific T-Cell Engager Is Active in Preclinical Models of Human Soft Tissue and Bone Sarcomas. Cancers 2023, 15, 1647.
- Tortorelli, I.; Navarria, F.; Di Maggio, A.; Banzato, A.; Lestuzzi, C.; Nicosia, L.; Chiusole, B.; Galiano, A.; Sbaraglia, M.; Zagonel, V.; et al. Trabectedin and Radiation Therapy for Cardiac Metastasis From Leiomyosarcoma: A Case Report and Review of the Literature. Front. Oncol. 2022, 12, 838114.
- Xie, W.; Forveille, S.; Iribarren, K.; Sauvat, A.; Senovilla, L.; Wang, Y.; Humeau, J.; Perez-Lanzon, M.; Zhou, H.; Martínez-Leal, J.F.; et al. Lurbinectedin Synergizes with Immune Checkpoint Blockade to Generate Anticancer Immunity. Oncoimmunology 2019, 8, e1656502.
- Cortesi, L.; Venturelli, M.; Barbieri, E.; Baldessari, C.; Bardasi, C.; Coccia, E.; Baglio, F.; Rimini, M.; Greco, S.; Napolitano, M.; et al. Exceptional Response to Lurbinectedin and Irinotecan in BRCA-Mutated Platinum-Resistant Ovarian Cancer Patient: A Case Report. Ther. Adv. Chronic Dis. 2022, 13, 20406223211063024.
- Takahashi, R.; Mabuchi, S.; Kawano, M.; Sasano, T.; Matsumoto, Y.; Kuroda, H.; Kozasa, K.; Hashimoto, K.; Sawada, K.; Kimura, T. Preclinical Investigations of PM01183 (Lurbinectedin) as a Single Agent or in Combination with Other Anticancer Agents for Clear Cell Carcinoma of the Ovary. PLoS ONE 2016, 11, e0151050.
- Schultz, C.W.; Zhang, Y.; Elmeskini, R.; Zimmermann, A.; Fu, H.; Murai, Y.; Wangsa, D.; Kumar, S.; Takahashi, N.; Atkinson, D.; et al. ATR Inhibition Augments the Efficacy of Lurbinectedin in Small-cell Lung Cancer. EMBO Mol. Med. 2023, 15, e17313.
- Porto, C.M.; Silva, V.D.L.; da Luz, J.S.B.; Filho, B.M.; da Silveira, V.M. Association between Vitamin D Deficiency and Heart Failure Risk in the Elderly. ESC Heart Fail. 2018, 5, 63–74.
- Poveda, A.; Lopez-Reig, R.; Oaknin, A.; Redondo, A.; Rubio, M.J.; Guerra, E.; Fariñas-Madrid, L.; Gallego, A.; Rodriguez-Freixinos, V.; Fernandez-Serra, A.; et al. Phase 2 Trial (POLA Study) of Lurbinectedin plus Olaparib in Patients with Advanced Solid Tumors: Results of Efficacy, Tolerability, and the Translational Study. Cancers 2022, 14, 915.
- Calvo, E.; Sessa, C.; Harada, G.; de Miguel, M.; Kahatt, C.; Luepke-Estefan, X.E.; Siguero, M.; Fernandez-Teruel, C.; Cullell-Young, M.; Stathis, A.; et al. Phase I Study of Lurbinectedin in Combination with Weekly Paclitaxel with or without Bevacizumab in Patients with Advanced Solid Tumors. Investig. New Drugs 2022, 40, 1263–1273.
- Metaxas, Y.; Früh, M.; Eboulet, E.I.; Grosso, F.; Pless, M.; Zucali, P.A.; Ceresoli, G.L.; Mark, M.; Schneider, M.; Maconi, A.; et al. Lurbinectedin as Second- or Third-Line Palliative Therapy in Malignant Pleural Mesothelioma: An International, Multi-Centre, Single-Arm, Phase II Trial (SAKK 17/16). Ann. Oncol. 2020, 31, 495–500.
- Metaxas, Y.; Cathomas, R.; Mark, M.; von Moos, R. Combination of Cisplatin and Lurbinectedin as Palliative Chemotherapy in Progressive Malignant Pleural Mesothelioma: Report of Two Cases. Lung Cancer 2016, 102, 136–138.
- Paz-Ares, L.; Forster, M.; Boni, V.; Szyldergemajn, S.; Corral, J.; Turnbull, S.; Cubillo, A.; Teruel, C.F.; Calderero, I.L.; Siguero, M.; et al. Phase I Clinical and Pharmacokinetic Study of PM01183 (a Tetrahydroisoquinoline, Lurbinectedin) in Combination with Gemcitabine in Patients with Advanced Solid Tumors. Investig. New Drugs 2017, 35, 198–206.
- Awada, A.H.; Boni, V.; Moreno, V.; Aftimos, P.; Kahatt, C.; Luepke-Estefan, X.E.; Siguero, M.; Fernandez-Teruel, C.; Cullell-Young, M.; Tabernero, J. Antitumor Activity of Lurbinectedin in Combination with Oral Capecitabine in Patients with Metastatic Breast Cancer. ESMO Open 2022, 7, 100651.
- Kristeleit, R.; Moreno, V.; Boni, V.; Guerra, E.M.; Kahatt, C.; Romero, I.; Calvo, E.; Basté, N.; López-Vilariño, J.A.; Siguero, M.; et al. Doxorubicin plus Lurbinectedin in Patients with Advanced Endometrial Cancer: Results from an Expanded Phase i Study. Int. J. Gynecol. Cancer 2021, 31, 1428–1436.
- Aix, S.P.; Ciuleanu, T.E.; Navarro, A.; Cousin, S.; Bonanno, L.; Smit, E.F.; Chiappori, A.; Olmedo, M.E.; Horvath, I.; Grohé, C.; et al. Combination Lurbinectedin and Doxorubicin versus Physician’s Choice of Chemotherapy in Patients with Relapsed Small-Cell Lung Cancer (ATLANTIS): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet. Respir. Med. 2023, 11, 74–86.
- Kim, K.H.; Kim, J.O.; Park, J.Y.; Seo, M.D.; Park, S.G. Antibody-Drug Conjugate Targeting c-Kit for the Treatment of Small Cell Lung Cancer. Int. J. Mol. Sci. 2022, 23, 2264.
- Digklia, A.; Coukos, G.; Homicsko, K. Trabectedin and Durvalumab Combination Is Feasible and Active in Relapsing Ovarian Cancer. Clin. Cancer Res. 2022, 28, 1745–1747.
- Ratti, C.; Botti, L.; Cancila, V.; Galvan, S.; Torselli, I.; Garofalo, C.; Manara, M.C.; Bongiovanni, L.; Valenti, C.F.; Burocchi, A.; et al. Trabectedin Overrides Osteosarcoma Differentiative Block and Reprograms the Tumor Immune Environment Enabling Effective Combination with Immune Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 5149–5161.
- Capasso Palmiero, U.; Morosi, L.; Bello, E.; Ponzo, M.; Frapolli, R.; Matteo, C.; Ferrari, M.; Zucchetti, M.; Minoli, L.; De Maglie, M.; et al. Readily Prepared Biodegradable Nanoparticles to Formulate Poorly Water Soluble Drugs Improving Their Pharmacological Properties: The Example of Trabectedin. J. Control. Release 2018, 276, 140–149.
More