Stroke remains one of the leading causes of death and disability worldwide. Current reperfusion treatments for ischaemic stroke are limited due to their narrow therapeutic window in rescuing ischaemic penumbra. Stem cell therapy offers a promising alternative. As a regenerative medicine, stem cells offer a wider range of treatment strategies, including long-term intervention for chronic patients, through the reparation and replacement of injured cells via mechanisms of differentiation and proliferation.
- stem cells
- cell-based therapy
- endothelial progenitor cells
- ischaemic stroke
1. Introduction
2. Pathology of Ischaemic Stroke
2.1. Excitotoxic Cell Death
The hypoxia that occurs in the immediate aftermath of an ischaemic attack triggers excitotoxic cell death. Hypoxic conditions downregulate ATP production by inhibiting plasma membrane Na+/K+/ATPase and Ca2+/ATPase pumps [7,8][7][8]. Receptor malfunction increases intracellular Na+ and Ca2+, causing cellular depolarisation and the propagation of action potentials. Na+ influx results in K+ efflux, further stimulating peri-infarct depolarisation. High levels of intracellular Ca2+ trigger glutamate exocytosis into the synaptic cleft; this accretion stimulates postsynaptic glutamate receptors, further increasing intracellular Ca2+ in the postsynaptic neurone [9,10][9][10]. An excessive Ca2+ load results in mitochondrial dysfunction, stimulating proteolysis and NADPH oxidase enzyme induction, triggering oxidative stress accompanied by the excessive release of reactive oxygen species (ROS). Once generated, ROS promote inflammatory mechanisms by attracting cytokines and leukocytes to infiltrate the brain as the BBB degrades [11,12,13,14][11][12][13][14]. Microglial cells, which are activated under oxidative stress, along with cytokines, also recruit matrix metalloproteinases (MMPs), a family of protease enzymes, further aiding local inflammation of ischaemic tissue [15]. Both activated microglia and reactive astrocytes are major components of the immune system in the brain, and the crosstalk between them reinforces the release of several proinflammatory factors, including IL-1β, IL-6, TNF-α, IL-15, and MMPs [16,17][16][17]. This homeostatic upset, inflammation, and uncontrolled enzymatic degradation inevitably damages the cellular structure and function and adversely affects the surrounding microenvironment.2.2. Apoptosis, Necrosis, and Necroptosis Pathways
A lack of Ca2+ homeostasis also stimulates numerous cellular death pathways. Ischaemia induces apoptosis via the release of cytochrome C from dysfunctional mitochondria followed by the activation of caspase-3 and the downstream hydrolases [18]. The cell enters the execution phase of apoptosis; the cytoplasm begins to shrink and display cytomorphological changes, including nuclear condensation [19]. Alternatively, the cell may undergo necrosis. This is often described as premature cell death and occurs due to Na+ influx accompanying Na+/K+ pump and Ca2+/ATP pump failure. Intracellular Na+ and Ca2+ aggregation leads to cellular oedema, swelling, and loss of lysosomal membrane integrity and cell rupture. Exposed cellular components attract digestive molecules for cell lysis, further contributing to local inflammation. It is noteworthy that unlike apoptosis, necrosis is independent of caspase activity [20,21][20][21].3. Blood–Brain Barrier
The BBB, an integral component of the neurovascular unit, regulates the selective passage of compounds between the blood and the brain parenchyma [15,25][15][22]. The BBB consists of pericytes, astrocytes, and endothelial cells (ECs) and is paracellularly sealed by tight junctions (TJs). These protein complexes are primarily composed of the transmembrane proteins claudins, occludins, junction adhesion molecules (JAMs), and zone occludens (ZO), an accessory protein responsible for manoeuvring cytoskeletal interactions [26][23]. The claudin family demonstrate a variety of transmembrane domains, of which the claudin-5 isoform is most greatly expressed, showing direct responsibility in tightening the BBB against small molecules (<800 Da) [26,27][23][24]. Occludins form dimers and oligomers, aiding paracellular permeability and stabilising barrier function, to which JAMs provide further support. The degradation of these tight junction constituents, catalysed by activated MMPs, compromises the BBB. Hypoxia and exaggerated local cytokine availability augment MMP expression, with elevated MMP-2 and MMP-9 levels identified in stroke patients [15,28,29,30,31,32][15][25][26][27][28][29]. The restoration of BBB integrity during the post-ischaemic period by MMP inhibition highlights this relationship [33,34][30][31]. The destruction of the basement membrane is another pathology to consider in ischaemic stroke. The basement membrane is a non-cellular complex consisting of a sheath of extracellular matrix and a series of proteins, namely collagen IV, nidogen, perlecan, agrin, and laminin. Although how the basement membrane becomes damaged during ischaemic stroke remains vague, it is presumed that the membrane undergoes dissolution, thereby exacerbating the loss of BBB and vascular integrity [37,38,39][32][33][34].4. Stem Cell as Therapeutics
A literature search using the key MeSH terms “stem cells”, “ischaemic stroke”, “stroke pathology”, “mesenchymal stem cells” (MSCs), “endothelial progenitor cells” (EPCs), “haematopoietic stem cells” (HSCs), and “neural stem cells” (NSCs) on the PubMed database identified relevant studies. Nottingham University search and Google Scholar were also used to collect pertinent studies. The mechanisms of stem cell therapy in ischaemic stroke is summarised in Figure 1.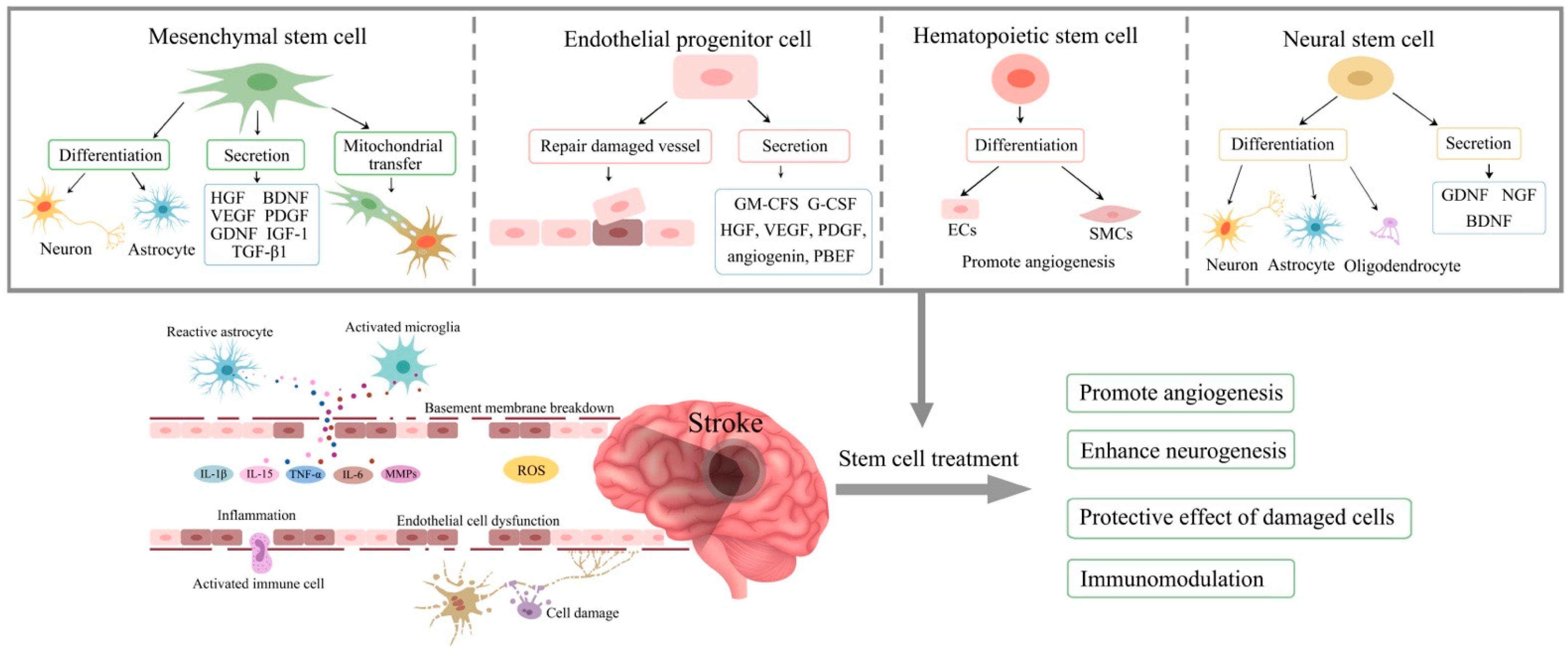
5. Mesenchymal Stem Cells
6. Endothelial Progenitor Cells (EPCs)
EPCs are circulating stem cells of endothelial origin. They migrate and accumulate in areas of vascular injury to help repair damaged vasculature through both neovascularisation and vascular remodelling. Due to their ability to detect and replace the damaged cerebral endothelial cells and restore BBB integrity by differentiating into mature endothelial cells, they are regarded as an important therapeutic for the management of ischaemic stroke [77,78][61][62]. An insufficient number and dysfunction of EPCs impairs vascular homeostasis and accelerates vascular disease [79,80][63][64]. EPCs are released into circulation by bone marrow in response to an ischaemic injury. They are isolated from the mononuclear cell (MNC) population through the use of specific antigens targeting endothelial cell maturity (e.g., KDR+), immaturity (e.g., CD133+), and stemness (e.g., CD34+) amongst a non-haematopoietic cell (CD45-) population [81][65]. To obtain cells that can be used for therapeutic purposes, MNCs are cultivated using specific endothelial cell media supplemented with a range of factors, including fibroblast growth factor (FGF), VEGF, insulin-like growth factor (IGF), hydrocortisone, ascorbic acid, and heparin [82,83,84][66][67][68]. The exogenous addition of EPCs repairs the integrity of an in vitro BBB model under OGD conditions and attenuates ischaemia-evoked oxidative stress and the apoptosis of endothelial cells [77,85,86][61][69][70]. EPCs in culture produce two distinct types of cells: early EPCs (eEPCs) and outgrowth ECs (OECs) or endothelial colony-forming cells (ECFCs). eEPCs represent an immature (CD133+) population of EPCs, with little proliferative capacity, appearing early in culture (three to four days). In contrast, OECs appear late in culture (two to four weeks) and demonstrate maturity and commitment to differentiation [93][71]. eEPCs and OECs can also be distinguished by their different morphology in that while eEPCs show a spindle-shaped morphology, OECs manifest the classical endothelial phenotype of cobblestone morphology [81,88][65][72]. A variety of agents, including VEGF, NO, EPO, SDF-1, and active MMP-9, regulate the mobilisation of EPCs from the BM into circulation [96,97][73][74]. VEGF, a key mediator of angiogenesis, stimulates EC proliferation, migration, and tube formation, eventually giving rise to new blood vessels and capillary networks [98,99,100][75][76][77]. The trafficking of EPCs, co-ordinated by SDF-1, is supported by the results of another in vivo investigation looking at the relationship between hypoxia-inducible factor-1 (HIF-1) and SDF-1 in ischaemic mice [29][26]. The study concluded that HIF-1 directly regulated SDF-1 gene expression in ischaemic tissue and that the migration and adhesion of EPCs to sites of injury was supported via CXCR4 and SDF-1 binding. Nitric oxide generated in endothelial cells by endothelial nitric oxide synthase (eNOS) is another important molecule that co-ordinates EPC proliferation and migration and inhibits apoptosis and platelet aggregation [103,104][78][79]. Observation of an impaired ischaemia-induced neovascularisation in eNOS-deficient mice bestows a key role on NO in mobilising EPCs. By inducing the phosphorylation of eNOS, VEGF plays an important role in stimulating NO production, a relationship confirmed by increases in the peripheral EPC count in normal mice after VEGF administration but not in eNOS-deficient mice [105][80]. Another pathway linked to the neovascular effects of EPCs is Notch1, a transmembrane receptor. Notch1 and its ligand Jagged1 have been implicated in post-ischaemic neovascularisation in both experimental and clinical stroke, where increases in the expression of activated Notch1 (Notch intracellular domain or NICD) in peri-infarct endothelial cells are coupled with the level of angiogenesis [107][81]. Neo-angiogenesis occurs by the proliferative sprouting of endothelial tip cells, followed and stabilised by endothelial stalk cells. Notch1 signalling co-ordinates this motility between tip and stalk cells and possibly directs arterial EC differentiation [108][82]. This relationship is supported by the suppression of tumour growth via the inhibition of Notch signalling [109][83].7. Haematopoietic Stem Cells
HSCs are multipotent, tissue-specific stem cells able to give rise to all functional blood cell types, including leukocytes, erythrocytes, and thrombocytes. HSCs present treatment possibilities as their supplementation encourages the recovery of diseased tissue by restoring blood and oxygen flow. The regeneration of ischaemic cells is facilitated by HSC differentiation (haematopoiesis), a process regulated by several hormones and cytokines, namely EPO, IL-3, granulocyte colony-stimulating factor (G-CSF), and macrophage colony-stimulating factor (M-CSF) [111][84]. CD34, though a surface marker expressed by other cells, is generally understood to represent hematopoietic stem and hematopoietic progenitor cells [112][85]. CD45 is another notable marker of HSCs [113][86]. The human adult produces over two hundred billion red blood cells per day [115][87]. With such a high turnover rate, the proliferative abilities of stem cells are most vitally exercised here where cell fate, regarding self-renewal or differentiation, is determined by gene expression and regulated by transcriptional factors [116,117][88][89]. Regulators between the two pathways are not distinct or separate, with factors able to influence cell fate down either route. However, some lineage-specific growth factors, such as G-CSF, M-CSF, and EPO, are categorical in directing HSCs down their respective pathways [118][90]. At high concentrations, GATA-1 suppresses the HSC exosome complex, consequently arresting early erythroblast proliferation and thus allowing for their maturation [119][91]. The Wnt and Notch pathways are other regulators of haematopoietic cell fate. Both Wnt and Notch receptors are widely expressed throughout the haematopoietic system and are critical in co-ordinating the development of leukocytes and their divisions. Wnt3a and Notch signalling promote early T-cell differentiation in human umbilical cord (hUCB) blood stem cells. Conversely, the inhibition of Wnt in the presence of Notch instead directs HSCs to give rise to natural killer cells [121][92]. Aside from transcriptional signalling, external situations also drive haematopoietic cell fate. For example, erythropoiesis occurs when HIF is activated under oxidative stress [122][93]. A study with MCAO rats provided insight into this relationship, where rats which intracerebrally received a culture of hypoxia-exposed (3% O2) HSCs displayed significantly better neurological outcomes compared to those which received normoxia-exposed (20% O2) or no treatment at all. This study also showed the role of exchange protein Epac1 in regulating the HIF/MMP pathway, with evidence connecting this communication to the promotion of neural progenitor cell (NPC) homing, aiding cerebral neuroplasticity. These results confirm previous findings documenting Epac1 action to enhance MMP activity and promote neovascularisation through the integrin-mediated adhesion of circulating HSCs to endothelial layers [123][94]. CD45+ bone marrow mononuclear cells (BMMNCs) were shown to differentiate into endothelial cells and smooth muscle cells to promote angiogenesis in an ischaemic stroke rat model [124][95].8. Neural Stem Cells
NSCs are undifferentiated stem cells of the CNS. They are multipotent stem cells able to self-renew and proliferate, give rise to different cell types, and differentiate into the three cell types of neural lineage, neurones, astrocytes, and oligodendrocytes [136][96]. Neurones, simply, are electrically excitable cells that synaptically transmit signals throughout the body [137][97]. Glial cells support and define these communications and are categorised by their functions; astrocytes maintain an appropriate chemical environment for brain functionality, and oligodendrocytes are responsible for myelination [138][98]. NSCs are sometimes referred to in the literature as “NPCs”, “neural precursor cells”, or “radial glia”, terminology which is used interchangeably and tends to be a difference in semantics. NSCs originate from the neuroectodermal tissue of the neural plate and are primarily found in the ventricular–subventricular zone (V-SVZ) of the walls of the lateral ventricles and the subgranular zone (SGZ) of the dentate nucleus [139,140][99][100]. NSCs are isolated by the enzymatic digestion of these locations [141][101] and quantified either in vitro using Reynolds and Weiss’ method of Neurosphere assay or by using a more recently developed collagen-based assay, Neural Colony-Forming Cell (NCFC) assay [142][102]. NCFC assays are now more commonly used as they are efficient in multiplying NSC count and can also discriminate between NSC and NPC populations by analysing the sizes of the colonies, representative of their proliferative abilities, the assay produces [143][103]. Neurogenesis is the growth and development of neuronal tissue and occurs both prenatally and in adults. It is the process by which NSCs develop into either neurones or glial cells (gliogenesis) and is influenced by both internal and external factors. Extrinsic factors in the local microenvironment of the SVZ and SGZ determine the lineage of NSCs, with soluble factors and transcriptional factors controlling intracellular signalling cascades such as the Notch-Hes1 pathway [144,145,146][104][105][106]. The activation of such pathways, triggered by oxidative pressures, decides whether NSCs will transform into astrocytes and oligodendrocytes or differentiate into neurones. By replacing necrotic neurones and positively influencing neuroregenerative pathways adversely affected by ischaemia, NSCs, through neurogenesis, present an exciting therapeutic option. The migration and differentiation of NSCs into mature neurons have been shown to restore cerebral homeostasis in MCAO rats [147][107]. Other therapeutic actions of NSCs, such as those including the modulation of the immunomodulatory response, reorganisation of neuronal pathways, and angiogenesis, somewhat resemble that of MSCs. The immunomodulatory properties of NSCs are supported by a marked attenuation in BBB damage, reduced cytokine production, and expression of proinflammatory markers IL-6 and TNF-α observed in acute stroke mice injected with a mixture of human-induced pluripotent stem cells (iPSCs) and NSCs in the hippocampus [148][108]. The behavioural improvements observed in these mice were comparable to those noted by other studies [149,150][109][110]. The Pilot Investigation of Stem Cells in Stroke (PISCES) trials are a collection of clinical studies looking at NSC treatment for ischaemic stroke. In response to a successful pre-clinical trial in which CTX-DP (a manufactured product as a suspension composed of CTX0E03 cells at a concentration of 5 × 104 cells/μL) yielded sensorimotor improvements in MCAO rats, an outsetting phase-I, open-label, dose-escalation study into the safety and tolerability of CTX-DP was conducted in human stroke patients [154][111]. The trial was thorough in its endeavours, analysing eleven men at a range of doses (three patients receiving two million CTX0E03 NSCs; three other patients receiving five million; three others receiving ten million; two others receiving twenty million) at a mean time of twenty-nine months (range from 6 to 60 months) after stroke onset.9. Route, Dose, and Timing of Treatment
9.1. Route
The main routes for treatment are IV, IA, and intracerebral administration. Both the pre-clinical and clinical studies show the IV route as the most preferred route due largely to its ease of use and its non-invasive nature. However, the IV treatment poses issues with engraftment and therapeutic efficacy due to the clearance of most cells by the lungs and liver during circulation [167,168][112][113]. IA administration is similar in technique as a minimally invasive and straightforward procedure. It is argued the IA route is more efficient than IV transport as this route does not lead to excessive cell trapping. While some studies comparing routes of stem cell delivery favour the IA route over IV administration [169][114], others report that there is no real difference in efficiency, with both routes revealing similar biodistribution rates and comparable functional outcomes [170,171,172][115][116][117]. Another route mentioned is the intracerebral route. The direct administration to site of injury eliminates the need to rely on chemical paracrine signalling in directing stem cell migration, allowing for smaller dose deliveries. This also, in theory, makes it a better option for chronic stroke patients (where the homing of stem cells may be weaker due to the absence of inflammatory mediators attracting as such) to maximise stem cell transfer; however, not all targets are physically accessible9.2. Dose
Despite the investigation of a wide range of cell concentrations in various clinical and pre-clinical studies, the optimal dose for an effective therapy after a cerebral ischaemic event continues to be a matter of debate. The lack of AEs at all doses tested negates the concerns regarding the numbers of stem cells to be administered and suggests the consideration of the reported efficacy of cells at a particular dose for a particular stem cell type. Though no clinical studies specifically evaluate the differences in stem cell efficacy at different concentrations, several studies comment on the safety over a range of cell doses. In tThe studies discussed in this paper, the ddoses of cells administered varied from 0.5 × 105 cells/kg to 6.1 × 108 cells [69,164][56][118].9.3. Timing
One of the biggest arguments for investing time and resources into stem cell research is the hope that the emerging treatment option(s) will demonstrate a larger therapeutic window than the current time limitations. The short life span of rodents is an issue when considering long-term intervention, explaining why pre-clinical studies fail to produce data on optimal treatment timing. Clinical trials, however, can evaluate the safety, feasibility, and efficacy of treatments with stem cells over a significant period of time. In addition, the time of administration varies significantly in clinical studies, ranging from twelve hours to twenty years, where safety is confirmed throughout [70,127][119][120]. There is little clinical evidence as to the application of stem cells during the hyperacute phase of stroke, so it is difficult to establish a consensus on the optimal timing of treatment in the immediate aftermath of stroke. In contrast, several clinical studies with acute stroke patients exist. They unanimously show that patients who received stem cells 7–72 h after stroke onset displayed better neurological outcomes [127,131][120][121].9.4. Comparison of Treatments with Different Types of Stem Cells
It is likely that treatments with different types of stem cells may yield different effects in the same disease settings which, in some cases, may be complementary. At present, not many clinical studies, if any, comparatively assess the therapeutic impact and safety of different stem cells in the same patient group. Future studies specifically exploring this issue in ischaemic stroke patients are likely to provide invaluable information as to the efficacy of different stem cells. They may also provide additional information about the dose and timing of administration of different stem cells.10. Conclusions
In conclusion, stem cell treatment presents possibilities for patients with all types of ischaemic stroke. With evidence of safety and efficacy measured in patients with acute, subacute, and chronic disease, therapeutic interventions appear to be promising for patients at every stage of the disease. However, further clinical research is necessary to standardise the treatment regimens.
References
- Brine, S. New Figures Show Larger Proportion of Strokes in the Middle Aged. 2018. Available online: https://www.gov.uk/government/news/new-figures-show-larger-proportion-of-strokes-in-the-middle-aged (accessed on 22 October 2023).
- Clark, W.M.; Albers, G.W.; Madden, K.P.; Hamilton, S. The rtPA (alteplase) 0- to 6-hour acute stroke trial, part A (A0276 g): Results of a double-blind, placebo-controlled, multicenter study. Thromblytic therapy in acute ischemic stroke study investigators. Stroke 2000, 31, 811–816.
- Del Zoppo, G.J.; Saver, J.L.; Jauch, E.C.; Adams, H.P., Jr.; American Heart Association Stroke Council. Expansion of the time window for treatment of acute ischemic stroke with intravenous tissue plasminogen activator: A science advisory from the American Heart Association/American Stroke Association. Stroke 2009, 40, 2945–2948.
- Broocks, G.; Kniep, H.; Kemmling, A.; Flottmann, F.; Nawabi, J.; Elsayed, S.; Schön, G.; Thomalla, G.; Fiehler, J.; Hanning, U. Effect of intravenous alteplase on ischaemic lesion water homeostasis. Eur. J. Neurol. 2020, 27, 376–383.
- Nogueira, R.G.; Jadhav, A.P.; Haussen, D.C.; Bonafe, A.; Budzik, R.F.; Bhuva, P.; Yavagal, D.R.; Ribo, M.; Cognard, C.; Hanel, R.A.; et al. Thrombectomy 6 to 24 Hours after Stroke with a Mismatch between Deficit and Infarct. N. Engl. J. Med. 2018, 378, 11–21.
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. 2018 Guidelines for the Early Management of Patients with Acute Ischemic Stroke: A Guideline for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2018, 49, e46–e110.
- Wheaton, W.W.; Chandel, N.S. Hypoxia. 2. Hypoxia regulates cellular metabolism. Am. J. Physiol. Cell Physiol. 2011, 300, C385–C393.
- Losenkova, K.; Zuccarini, M.; Helenius, M.; Jacquemet, G.; Gerasimovskaya, E.; Tallgren, C.; Jalkanen, S.; Yegutkin, G.G. Endothelial cells cope with hypoxia-induced depletion of ATP via activation of cellular purine turnover and phosphotransfer networks. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1804–1815.
- Annunziato, L.; Cataldi, M.; Pignataro, G.; Secondo, A.; Molinaro, P. Glutamate-independent calcium toxicity: Introduction. Stroke 2007, 38, 661–664.
- Bano, D.; Nicotera, P. Ca2+ Signals and Neuronal Death in Brain Ischemia. Stroke 2007, 38, 674–676.
- Garcia, J.H.; Liu, K.F.; Yoshida, Y.; Lian, J.; Chen, S.; del Zoppo, G.J. Influx of leukocytes and platelets in an evolving brain infarct (Wistar rat). Am. J. Pathol. 1994, 144, 188–199.
- Gursoy-Ozdemir, Y.; Can, A.; Dalkara, T. Reperfusion-induced oxidative/nitrative injury to neurovascular unit after focal cerebral ischemia. Stroke 2004, 35, 1449–1453.
- Gibson, C.L.; Srivastava, K.; Sprigg, N.; Bath, P.M.; Bayraktutan, U. Inhibition of Rho-kinase protects cerebral barrier from ischaemia-evoked injury through modulations of endothelial cell oxidative stress and tight junctions. J. Neurochem. 2014, 129, 816–826.
- Rakkar, K.; Bayraktutan, U. Increases in intracellular calcium perturb blood-brain barrier via protein kinase C-α and apoptosis. Biochim. Biophys. Acta 2016, 1862, 56–71.
- Kim, K.-A.; Shin, D.; Kim, J.-H.; Shin, Y.-J.; Rajanikant, G.K.; Majid, A.; Baek, S.-H.; Bae, O.-N. Role of Autophagy in Endothelial Damage and Blood–Brain Barrier Disruption in Ischemic Stroke. Stroke 2018, 49, 1571–1579.
- Shi, S.X.; Li, Y.J.; Shi, K.; Wood, K.; Ducruet, A.F.; Liu, Q. IL (Interleukin)-15 Bridges Astrocyte-Microglia Crosstalk and Exacerbates Brain Injury Following Intracerebral Hemorrhage. Stroke 2020, 51, 967–974.
- He, T.; Yang, G.Y.; Zhang, Z. Crosstalk of Astrocytes and Other Cells during Ischemic Stroke. Life 2022, 12, 910.
- Broughton, B.R.; Reutens, D.C.; Sobey, C.G. Apoptotic mechanisms after cerebral ischemia. Stroke 2009, 40, e331–e339.
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516.
- Skulachev, V.P. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis 2006, 11, 473–485.
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48.
- Nian, K.; Harding, I.C.; Herman, I.M.; Ebong, E.E. Blood-Brain Barrier Damage in Ischemic Stroke and Its Regulation by Endothelial Mechanotransduction. Front. Physiol. 2020, 11, 605398.
- Winkler, L.; Blasig, R.; Breitkreuz-Korff, O.; Berndt, P.; Dithmer, S.; Helms, H.C.; Puchkov, D.; Devraj, K.; Kaya, M.; Qin, Z.; et al. Tight junctions in the blood–brain barrier promote edema formation and infarct size in stroke—Ambivalent effects of sealing proteins. J. Cereb. Blood Flow Metab. 2021, 41, 132–145.
- Nakano-Doi, A.; Sakuma, R.; Matsuyama, T.; Nakagomi, T. Ischemic stroke activates the VE-cadherin promoter and increases VE-cadherin expression in adult mice. Histol. Histopathol. 2018, 33, 507–521.
- Kalka, C.; Masuda, H.; Takahashi, T.; Gordon, R.; Tepper, O.; Gravereaux, E.; Pieczek, A.; Iwaguro, H.; Hayashi, S.-I.; Isner, J.M.; et al. Vascular Endothelial Growth Factor165 Gene Transfer Augments Circulating Endothelial Progenitor Cells in Human Subjects. Circ. Res. 2000, 86, 1198–1202.
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864.
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol. Cell Physiol. 2018, 315, C343–C356.
- Savitz, S.I.; Yavagal, D.; Rappard, G.; Likosky, W.; Rutledge, N.; Graffagnino, C.; Alderazi, Y.; Elder, J.A.; Chen, P.R.; Budzik, R.F.; et al. A Phase 2 Randomized, Sham-Controlled Trial of Internal Carotid Artery Infusion of Autologous Bone Marrow–Derived ALD-401 Cells in Patients with Recent Stable Ischemic Stroke (RECOVER-Stroke). Circulation 2019, 139, 192–205.
- Abdullah, Z.; Bayraktutan, U. Suppression of PKC-α attenuates TNF-α-evoked cerebral barrier breakdown via regulations of MMP-2 and plasminogen-plasmin system. Biochim. Biophys. Acta 2016, 1862, 1354–1366.
- Asahi, M.; Wang, X.; Mori, T.; Sumii, T.; Jung, J.C.; Moskowitz, M.A.; Fini, M.E.; Lo, E.H. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J. Neurosci. 2001, 21, 7724–7732.
- Fujimoto, M.; Takagi, Y.; Aoki, T.; Hayase, M.; Marumo, T.; Gomi, M.; Nishimura, M.; Kataoka, H.; Hashimoto, N.; Nozaki, K. Tissue inhibitor of metalloproteinases protect blood-brain barrier disruption in focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 1674–1685.
- Xu, L.; Nirwane, A.; Yao, Y. Basement membrane and blood-brain barrier. Stroke Vasc. Neurol. 2019, 4, 78–82.
- Yao, Y. Basement membrane and stroke. J. Cereb. Blood Flow Metab. 2019, 39, 3–19.
- Kang, M.; Yao, Y. Basement Membrane Changes in Ischemic Stroke. Stroke 2020, 51, 1344–1352.
- Krabbe, C.; Zimmer, J.; Meyer, M. Neural transdifferentiation of mesenchymal stem cells—A critical review. APMIS 2005, 113, 831–844.
- Wang, F.; Tang, H.; Zhu, J.; Zhang, J.H. Transplanting Mesenchymal Stem Cells for Treatment of Ischemic Stroke. Cell Transplant. 2018, 27, 1825–1834.
- Croft, A.P.; Przyborski, S.A. Mesenchymal stem cells expressing neural antigens instruct a neurogenic cell fate on neural stem cells. Exp. Neurol. 2009, 216, 329–341.
- Lian, Q.; Zhang, Y.; Liang, X.; Gao, F.; Tse, H.-F. Directed Differentiation of Human-Induced Pluripotent Stem Cells to Mesenchymal Stem Cells. In Mesenchymal Stem Cells: Methods and Protocols; Gnecchi, M., Ed.; Springer: New York, NY, USA, 2016; pp. 289–298.
- Khan, A.A.; Huat, T.J.; Al Mutery, A.; El-Serafi, A.T.; Kacem, H.H.; Abdallah, S.H.; Reza, M.F.; Abdullah, J.M.; Jaafar, H. Significant transcriptomic changes are associated with differentiation of bone marrow-derived mesenchymal stem cells into neural progenitor-like cells in the presence of bFGF and EGF. Cell Biosci. 2020, 10, 126.
- Kruminis-Kaszkiel, E.; Osowski, A.; Bejer-Oleńska, E.; Dziekoński, M.; Wojtkiewicz, J. Differentiation of Human Mesenchymal Stem Cells from Wharton’s Jelly Towards Neural Stem Cells Using a Feasible and Repeatable Protocol. Cells 2020, 9, 739.
- Venkat, P.; Shen, Y.; Chopp, M.; Chen, J. Cell-based and pharmacological neurorestorative therapies for ischemic stroke. Neuropharmacology 2018, 134, 310–322.
- Fu, X.; Liu, G.; Halim, A.; Ju, Y.; Luo, Q.; Song, G. Mesenchymal Stem Cell Migration and Tissue Repair. Cells 2019, 8, 784.
- Zou, C.; Luo, Q.; Qin, J.; Shi, Y.; Yang, L.; Ju, B.; Song, G. Osteopontin Promotes Mesenchymal Stem Cell Migration and Lessens Cell Stiffness via Integrin β1, FAK, and ERK Pathways. Cell Biochem. Biophys. 2013, 65, 455–462.
- Huang, P.; Gebhart, N.; Richelson, E.; Brott, T.G.; Meschia, J.F.; Zubair, A.C. Mechanism of mesenchymal stem cell-induced neuron recovery and anti-inflammation. Cytotherapy 2014, 16, 1336–1344.
- Maacha, S.; Sidahmed, H.; Jacob, S.; Gentilcore, G.; Calzone, R.; Grivel, J.-C.; Cugno, C. Paracrine Mechanisms of Mesenchymal Stromal Cells in Angiogenesis. Stem Cells Int. 2020, 2020, 4356359.
- Tai, L.; Saffery, N.S.; Chin, S.P.; Cheong, S.K. Secretome profile of TNF-α-induced human umbilical cord mesenchymal stem cells unveils biological processes relevant to skin wound healing. Regen. Med. 2023, 18, 839–856.
- Pankajakshan, D.; Agrawal, D.K. Mesenchymal Stem Cell Paracrine Factors in Vascular Repair and Regeneration. J. Biomed. Technol. Res. 2014, 1.
- Wang, J.; Fu, X.; Jiang, C.; Yu, L.; Wang, M.; Han, W.; Liu, L.; Wang, J. Bone marrow mononuclear cell transplantation promotes therapeutic angiogenesis via upregulation of the VEGF–VEGFR2 signaling pathway in a rat model of vascular dementia. Behav. Brain Res. 2014, 265, 171–180.
- Mu, J.; Bakreen, A.; Juntunen, M.; Korhonen, P.; Oinonen, E.; Cui, L.; Myllyniemi, M.; Zhao, S.; Miettinen, S.; Jolkkonen, J. Combined Adipose Tissue-Derived Mesenchymal Stem Cell Therapy and Rehabilitation in Experimental Stroke. Front. Neurol. 2019, 10, 235.
- Tsai, M.J.; Tsai, S.K.; Hu, B.R.; Liou, D.Y.; Huang, S.L.; Huang, M.C.; Huang, W.C.; Cheng, H.; Huang, S.S. Recovery of neurological function of ischemic stroke by application of conditioned medium of bone marrow mesenchymal stem cells derived from normal and cerebral ischemia rats. J. Biomed. Sci. 2014, 21, 5.
- Zang, J.; Sha, M.; Zhang, C.; Ye, J.; Zhang, K.; Gao, J. Senescent hepatocyte secretion of matrix metalloproteinases is regulated by nuclear factor-κB signaling. Life Sci. 2017, 191, 205–210.
- Bhasin, A.; Padma Srivastava, M.V.; Mohanty, S.; Bhatia, R.; Kumaran, S.S.; Bose, S. Stem cell therapy: A clinical trial of stroke. Clin. Neurol. Neurosurg. 2013, 115, 1003–1008.
- Honmou, O.; Houkin, K.; Matsunaga, T.; Niitsu, Y.; Ishiai, S.; Onodera, R.; Waxman, S.G.; Kocsis, J.D. Intravenous administration of auto serum-expanded autologous mesenchymal stem cells in stroke. Brain 2011, 134, 1790–1807.
- Lee, J.S.; Hong, J.M.; Moon, G.J.; Lee, P.H.; Ahn, Y.H.; Bang, O.Y.; STARTING Collaborators. A long-term follow-up study of intravenous autologous mesenchymal stem cell transplantation in patients with ischemic stroke. Stem Cells 2010, 28, 1099–1106.
- Steinberg, G.K.; Kondziolka, D.; Wechsler, L.R.; Lunsford, L.D.; Coburn, M.L.; Billigen, J.B.; Kim, A.S.; Johnson, J.N.; Bates, D.; King, B.; et al. Clinical Outcomes of Transplanted Modified Bone Marrow-Derived Mesenchymal Stem Cells in Stroke: A Phase 1/2a Study. Stroke 2016, 47, 1817–1824.
- Bhatia, V.; Gupta, V.; Khurana, D.; Sharma, R.R.; Khandelwal, N. Randomized Assessment of the Safety and Efficacy of Intra-Arterial Infusion of Autologous Stem Cells in Subacute Ischemic Stroke. Am. J. Neuroradiol. 2018, 39, 899–904.
- Han, H.; Hu, J.; Yan, Q.; Zhu, J.; Zhu, Z.; Chen, Y.; Sun, J.; Zhang, R. Bone marrow-derived mesenchymal stem cells rescue injured H9c2 cells via transferring intact mitochondria through tunneling nanotubes in an in vitro simulated ischemia/reperfusion model. Mol. Med. Rep. 2016, 13, 1517–1524.
- Tseng, N.; Lambie, S.C.; Huynh, C.Q.; Sanford, B.; Patel, M.; Herson, P.S.; Ormond, D.R. Mitochondrial transfer from mesenchymal stem cells improves neuronal metabolism after oxidant injury in vitro: The role of Miro1. J. Cereb. Blood Flow Metab. 2021, 41, 761–770.
- Yang, Y.; Ye, G.; Zhang, Y.-L.; He, H.-W.; Yu, B.-Q.; Hong, Y.-M.; You, W.; Li, X. Transfer of mitochondria from mesenchymal stem cells derived from induced pluripotent stem cells attenuates hypoxia-ischemia-induced mitochondrial dysfunction in PC12 cells. Neural Regen. Res. 2020, 15, 464–472.
- Noronha, N.d.C.; Mizukami, A.; Caliári-Oliveira, C.; Cominal, J.G.; Rocha, J.L.M.; Covas, D.T.; Swiech, K.; Malmegrim, K.C.R. Priming approaches to improve the efficacy of mesenchymal stromal cell-based therapies. Stem Cell Res. Ther. 2019, 10, 131.
- Kadir, R.R.A.; Alwjwaj, M.; Bayraktutan, U. Treatment with outgrowth endothelial cells protects cerebral barrier against ischemic injury. Cytotherapy 2022, 24, 489–499.
- Bayraktutan, U. Endothelium, endothelial progenitor cells and stroke. J. Neurol. Clin. Neurosci. 2017, 1, 21–22.
- Williamson, K.; Stringer, S.E.; Alexander, M.Y. Endothelial progenitor cells enter the aging arena. Front. Physiol. 2012, 3, 30.
- Ya, J.; Bayraktutan, U. Vascular Ageing: Mechanisms, Risk Factors, and Treatment Strategies. Int. J. Mol. Sci. 2023, 24, 11538.
- Rakkar, K.; Othman, O.; Sprigg, N.; Bath, P.; Bayraktutan, U. Endothelial progenitor cells, potential biomarkers for diagnosis and prognosis of ischemic stroke: Protocol for an observational case-control study. Neural Regen. Res. 2020, 15, 1300–1307.
- Urbich, C.; Dimmeler, S. Endothelial progenitor cells: Characterization and role in vascular biology. Circ. Res. 2004, 95, 343–353.
- Yuan, J.-J.; Yang, J.; Sun, S.-L.; Zhang, R.; Xu, Y.-M. Endothelial Progenitor Cells’ Classification and Application in Neurological Diseases. Tissue Eng. Regen. Med. 2017, 14, 327–332.
- Bayraktutan, U. Endothelial progenitor cells: Potential novel therapeutics for ischaemic stroke. Pharmacol. Res. 2019, 144, 181–191.
- Kadir, R.R.A.; Alwjwaj, M.; Bayraktutan, U. Protein kinase C-beta distinctly regulates blood-brain barrier-forming capacity of Brain Microvascular endothelial cells and outgrowth endothelial cells. Metab. Brain Dis. 2022, 37, 1815–1827.
- Alwjwaj, M.; Kadir, R.R.A.; Bayraktutan, U. Outgrowth endothelial progenitor cells restore cerebral barrier function following ischaemic damage: The impact of NOX2 inhibition. Eur. J. Neurosci. 2022, 55, 1658–1670.
- Shmelkov, S.V.; Butler, J.M.; Hooper, A.T.; Hormigo, A.; Kushner, J.; Milde, T.; St Clair, R.; Baljevic, M.; White, I.; Jin, D.K.; et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133− metastatic colon cancer cells initiate tumors. J. Clin. Investig. 2008, 118, 2111–2120.
- Alwjwaj, M.; Kadir, R.R.A.; Bayraktutan, U. The secretome of endothelial progenitor cells: A potential therapeutic strategy for ischemic stroke. Neural Regen. Res. 2021, 16, 1483–1489.
- Hristov, M.; Erl, W.; Weber, P.C. Endothelial Progenitor Cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1185–1189.
- Liao, S.; Luo, C.; Cao, B.; Hu, H.; Wang, S.; Yue, H.; Chen, L.; Zhou, Z. Endothelial Progenitor Cells for Ischemic Stroke: Update on Basic Research and Application. Stem Cells Int. 2017, 2017, 2193432.
- Niu, G.; Chen, X. Vascular Endothelial Growth Factor as an Anti-Angiogenic Target for Cancer Therapy. Curr. Drug Targets 2010, 11, 1000–1017.
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105.
- Li, L.; Liu, H.; Xu, C.; Deng, M.; Song, M.; Yu, X.; Xu, S.; Zhao, X. VEGF promotes endothelial progenitor cell differentiation and vascular repair through connexin 43. Stem Cell Res. Ther. 2017, 8, 237.
- Förstermann, U.; Münzel, T. Endothelial Nitric Oxide Synthase in Vascular Disease. Circulation 2006, 113, 1708–1714.
- Lei, J.; Vodovotz, Y.; Tzeng, E.; Billiar, T.R. Nitric oxide, a protective molecule in the cardiovascular system. Nitric Oxide 2013, 35, 175–185.
- Feliers, D.; Chen, X.; Akis, N.; Choudhury, G.G.; Madaio, M.; Kasinath, B.S. VEGF regulation of endothelial nitric oxide synthase in glomerular endothelial cells. Kidney Int. 2005, 68, 1648–1659.
- Ren, C.; Yao, Y.; Han, R.; Huang, Q.; Li, H.; Wang, B.; Li, S.; Li, M.; Mao, Y.; Mao, X.; et al. Cerebral ischemia induces angiogenesis in the peri-infarct regions via Notch1 signaling activation. Exp. Neurol. 2018, 304, 30–40.
- Akil, A.; Gutiérrez-García, A.K.; Guenter, R.; Rose, J.B.; Beck, A.W.; Chen, H.; Ren, B. Notch Signaling in Vascular Endothelial Cells, Angiogenesis, and Tumor Progression: An Update and Prospective. Front. Cell Dev. Biol. 2021, 9, 642352.
- Mack, J.J.; Iruela-Arispe, M.L. NOTCH regulation of the endothelial cell phenotype. Curr. Opin. Hematol. 2018, 25, 212–218.
- Smith, B.R. Regulation of hematopoiesis. Yale J. Biol. Med. 1990, 63, 371–380.
- Sidney, L.E.; Branch, M.J.; Dunphy, S.E.; Dua, H.S.; Hopkinson, A. Concise Review: Evidence for CD34 as a Common Marker for Diverse Progenitors. Stem Cells 2014, 32, 1380–1389.
- Ngo, N.; Patel, K.; Isaacson, P.G.; Naresh, K.N. Leucocyte common antigen (CD45) and CD5 positivity in an “undifferentiated” carcinoma: A potential diagnostic pitfall. J. Clin. Pathol. 2007, 60, 936–938.
- Britannica, T.; Editors of Encyclopaedia. organ. Encyclopedia Britannica. 22 August 2023. Available online: https://www.britannica.com/science/organ-biology (accessed on 22 October 2023).
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An Evolving Paradigm for Stem Cell Biology. Cell 2008, 132, 631–644.
- Mayani, H. The regulation of hematopoietic stem cell populations . F1000Research 2016, 5, 1524.
- Rodrigues, N.P.; Tipping, A.J.; Wang, Z.; Enver, T. GATA-2 mediated regulation of normal hematopoietic stem/progenitor cell function, myelodysplasia and myeloid leukemia. Int. J. Biochem. Cell Biol. 2012, 44, 457–460.
- McIver, S.C.; Kang, Y.-A.; DeVilbiss, A.W.; O’Driscoll, C.A.; Ouellette, J.N.; Pope, N.J.; Camprecios, G.; Chang, C.-J.; Yang, D.; Bouhassira, E.E.; et al. The exosome complex establishes a barricade to erythroid maturation. Blood 2014, 124, 2285–2297.
- Aoyama, K.; Delaney, C.; Varnum-Finney, B.; Kohn, A.D.; Moon, R.T.; Bernstein, I.D. The Interaction of the Wnt and Notch Pathways Modulates Natural Killer Versus T Cell Differentiation. Stem Cells 2007, 25, 2488–2497.
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53.
- Lin, C.-H.; Lee, H.-T.; Lee, S.-D.; Lee, W.; Cho, C.-W.C.; Lin, S.-Z.; Wang, H.-J.; Okano, H.; Su, C.-Y.; Yu, Y.-L.; et al. Role of HIF-1α-activated Epac1 on HSC-mediated neuroplasticity in stroke model. Neurobiol. Dis. 2013, 58, 76–91.
- Wang, J.; Yu, L.; Jiang, C.; Chen, M.; Ou, C.; Wang, J. Bone marrow mononuclear cells exert long-term neuroprotection in a rat model of ischemic stroke by promoting arteriogenesis and angiogenesis. Brain Behav. Immun. 2013, 34, 56–66.
- Kennea, N.L.; Mehmet, H. Neural stem cells. J. Pathol. 2002, 197, 536–550.
- Ludwig, P.E.; Reddy, V.; Varacallo, M. Neuroanatomy, Neurons. . In StatPearls ; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK441977/ (accessed on 22 October 2023).
- Purves, D.; Augustine, G.J.; Fitzpatrick, D.; Katz, L.C.; Lamantia, A.; McNamara, J.O.; Williams, S.M. (Eds.) Neuroscience, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2001. Available online: https://www.ncbi.nlm.nih.gov/books/NBK10869/ (accessed on 22 October 2023).
- Martínez-Cerdeño, V.; Noctor, S.C. Neural Progenitor Cell Terminology. Front. Neuroanat. 2018, 12, 104.
- Obernier, K.; Alvarez-Buylla, A. Neural stem cells: Origin, heterogeneity and regulation in the adult mammalian brain. Development 2019, 146, dev156059.
- Ferrari, D.; Binda, E.; Filippis, L.D.; Vescovi, A.L. Isolation of Neural Stem Cells from Neural Tissues Using the Neurosphere Technique. Curr. Protoc. Stem Cell Biol. 2010, 15, 2D.6.1–2D.6.18.
- Reynolds, B.A.; Weiss, S. Generation of Neurons and Astrocytes from Isolated Cells of the Adult Mammalian Central Nervous System. Science 1992, 255, 1707–1710.
- Louis, S.A.; Mak, C.K.H. Enumerating Stem Cell Frequency: Neural Colony Forming Cell Assay. In Neural Progenitor Cells: Methods and Protocols; Reynolds, B.A., Deleyrolle, L.P., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 117–132.
- Zhou, Z.-D.; Kumari, U.; Xiao, Z.-C.; Tan, E.-K. Notch as a molecular switch in neural stem cells. IUBMB Life 2010, 62, 618–623.
- Faigle, R.; Song, H. Signaling mechanisms regulating adult neural stem cells and neurogenesis. Biochim. Biophys. Acta BBA Gen. Subj. 2013, 1830, 2435–2448.
- Vieira, M.S.; Santos, A.K.; Vasconcellos, R.; Goulart, V.A.M.; Parreira, R.C.; Kihara, A.H.; Ulrich, H.; Resende, R.R. Neural stem cell differentiation into mature neurons: Mechanisms of regulation and biotechnological applications. Biotechnol. Adv. 2018, 36, 1946–1970.
- Yuan, T.; Liao, W.; Feng, N.-H.; Lou, Y.-L.; Niu, X.; Zhang, A.-J.; Wang, Y.; Deng, Z.-F. Human induced pluripotent stem cell-derived neural stem cells survive, migrate, differentiate, and improve neurologic function in a rat model of middle cerebral artery occlusion. Stem Cell Res. Ther. 2013, 4, 73.
- Eckert, A.; Huang, L.; Gonzalez, R.; Kim, H.-S.; Hamblin, M.H.; Lee, J.-P. Bystander Effect Fuels Human Induced Pluripotent Stem Cell-Derived Neural Stem Cells to Quickly Attenuate Early Stage Neurological Deficits after Stroke. Stem Cells Transl. Med. 2015, 4, 841–851.
- Huang, L.; Wong, S.; Snyder, E.Y.; Hamblin, M.H.; Lee, J.-P. Human neural stem cells rapidly ameliorate symptomatic inflammation in early-stage ischemic-reperfusion cerebral injury. Stem Cell Res. Ther. 2014, 5, 129.
- Watanabe, T.; Nagai, A.; Sheikh, A.M.; Mitaki, S.; Wakabayashi, K.; Kim, S.U.; Kobayashi, S.; Yamaguchi, S. A human neural stem cell line provides neuroprotection and improves neurological performance by early intervention of neuroinflammatory system. Brain Res. 2016, 1631, 194–203.
- Kalladka, D.; Sinden, J.; Pollock, K.; Haig, C.; McLean, J.; Smith, W.; McConnachie, A.; Santosh, C.; Bath, P.M.; Dunn, L.; et al. Human neural stem cells in patients with chronic ischaemic stroke (PISCES): A phase 1, first-in-man study. Lancet 2016, 388, 787–796.
- Wechsler, L.R.; Bates, D.; Stroemer, P.; Andrews-Zwilling, Y.S.; Aizman, I. Cell Therapy for Chronic Stroke. Stroke 2018, 49, 1066–1074.
- Baker, E.W.; Kinder, H.A.; West, F.D. Neural stem cell therapy for stroke: A multimechanistic approach to restoring neurological function. Brain Behav. 2019, 9, e01214.
- Zhang, H.-L.; Xie, X.-F.; Xiong, Y.-Q.; Liu, S.-M.; Hu, G.-Z.; Cao, W.-F.; Wu, X.-M. Comparisons of the therapeutic effects of three different routes of bone marrow mesenchymal stem cell transplantation in cerebral ischemic rats. Brain Res. 2018, 1680, 143–154.
- Vasconcelos-dos-Santos, A.; Rosado-de-Castro, P.H.; Lopes de Souza, S.A.; da Costa Silva, J.; Ramos, A.B.; Rodriguez de Freitas, G.; Barbosa da Fonseca, L.M.; Gutfilen, B.; Mendez-Otero, R. Intravenous and intra-arterial administration of bone marrow mononuclear cells after focal cerebral ischemia: Is there a difference in biodistribution and efficacy? Stem Cell Res. 2012, 9, 1–8.
- Rosado-de-Castro, P.H.; Schmidt Fda, R.; Battistella, V.; Lopes de Souza, S.A.; Gutfilen, B.; Goldenberg, R.C.; Kasai-Brunswick, T.H.; Vairo, L.; Silva, R.M.; Wajnberg, E.; et al. Biodistribution of bone marrow mononuclear cells after intra-arterial or intravenous transplantation in subacute stroke patients. Regen. Med. 2013, 8, 145–155.
- Yang, B.; Migliati, E.; Parsha, K.; Schaar, K.; Xi, X.; Aronowski, J.; Savitz, S.I. Intra-Arterial Delivery Is Not Superior to Intravenous Delivery of Autologous Bone Marrow Mononuclear Cells in Acute Ischemic Stroke. Stroke 2013, 44, 3463–3472.
- Levy, M.L.; Crawford, J.R.; Dib, N.; Verkh, L.; Tankovich, N.; Cramer, S.C. Phase I/II Study of Safety and Preliminary Efficacy of Intravenous Allogeneic Mesenchymal Stem Cells in Chronic Stroke. Stroke 2019, 50, 2835–2841.
- Chen, L.; Xi, H.; Huang, H.; Zhang, F.; Liu, Y.; Chen, D.; Xiao, J. Multiple Cell Transplantation Based on an Intraparenchymal Approach for Patients with Chronic Phase Stroke. Cell Transplant. 2013, 22, 83–91.
- Boy, S.; Sauerbruch, S.; Kraemer, M.; Schormann, T.; Schlachetzki, F.; Schuierer, G.; Luerding, R.; Hennemann, B.; Orso, E.; Dabringhaus, A.; et al. Mobilisation of Hematopoietic CD34+ Precursor Cells in Patients with Acute Stroke Is Safe—Results of an Open-Labeled Non Randomized Phase I/II Trial. PLoS ONE 2011, 6, e23099.
- Savitz, S.I.; Misra, V.; Kasam, M.; Juneja, H.; Cox, C.S., Jr.; Alderman, S.; Aisiku, I.; Kar, S.; Gee, A.; Grotta, J.C. Intravenous autologous bone marrow mononuclear cells for ischemic stroke. Ann. Neurol. 2011, 70, 59–69.