2. Advantages of Using Yeast Models to Investigate Oncohistones
While much has been uncovered about oncohistones since their discovery about a decade ago
[3][4][5][61,62,63], many outstanding questions remain fully understand how the conversion from essential protein to oncogenic driver occurs. A major complication in the investigation of how an oncohistone alters histone function and growth patterns in human cells is the large number of histone genes. Engineering cell lines that faithfully recapitulate the genetic environment found in disease or that solely express mutant histone in the absence of wild type histone is quite technically challenging. While one recent study in mouse embryonic stem cells introduced an H3K27 mutant (H3K27R) into all 28 alleles of the genes encoding histone H3
[6][87], the engineering required for designing and generating the lines is not conducive to screening a large number of potential oncohistone mutations for functional changes. Therefore, organisms such as budding or fission yeast can circumvent the challenges posed by mammalian cell lines by having fewer histone genes and allow modeling of oncohistone mutations in a simpler genetic context where the histone variant can be readily expressed as the sole histone protein present in the cell.
As the majority of oncohistone studies have focused on histone H3, focusing on this core histone protein is of particular interest. Notably, in comparing yeast and human histone H3, the only residues in the N-terminal tail that are not strictly identical are conservative changes, meaning that the amino acids likely serve similar biochemical functions. This observation is crucial as the N-terminal tail is critical for the dynamic receipt and sending of epigenetic signals via the histone code. Not only is the protein sequence highly similar, but the 3-dimensional structures of human and yeast H3 proteins are also highly conserved
[7][25]. These similarities suggest that histone H3 performs analogous functions in each species, and thus, that molecular disruptions caused by the oncohistone mutants would be similar.
Importantly, many associated epigenetic factors are also highly conserved between humans and budding yeast
[8][9][10][88,89,90]. Given the dynamic and layered regulation that occurs in histones, findings uncovered in model organisms are greatly strengthened by the shared structures and functions with humans at various levels. For example, the human acetyl transferases Gcn5 and Tip60 and the methyl transferase SetD2 have conserved counterparts in yeast
[8][9][10][88,89,90]. A notable exception is the PRC2 complex, which methylates H3K27 in humans, and is not present in
S. cerevisiae or
S. pombe [11][91]. While many histone modifying enzymes are conserved, there are limitations regarding which ones can be investigated in yeast models as some mammalian proteins lack yeast orthologs. In addition to conserved associated enzymes, the functional consequences of many key PTMs are similar between yeast and mammals. For example, H3K36 methylation promotes double-strand break repair in budding yeast and humans
[12][13][14][82,92,93], and heterochromatin is marked by methylation at H3K9 in fission yeast and humans
[15][16][52,94]. There are differences, though, with both budding and fission yeast lacking H3K27 methylation
[17][95]. Thus, yeast systems can model the placement and consequence of many, though not all, epigenetic marks found in mammalian species.
The yeast model systems offer flexibility in how to analyze the functional consequences of oncohistone mutations by examining cells that express the oncohistone protein as the sole copy of the histone present. In a mammalian cell line, engineering the 15 copies of histone H3 to express the oncohistone as the sole histone protein for clean characterization of its impact is quite technically challenging, although this task has been accomplished for H3K27
[6][87]. However, in the budding yeast system, where each histone is encoded by only two genes, one can readily engineer cells that express an oncohistone as the sole histone protein present. In fact, offering flexibility, one copy of a histone gene can be altered using genome editing and then the other histone gene can be kept intact or deleted (
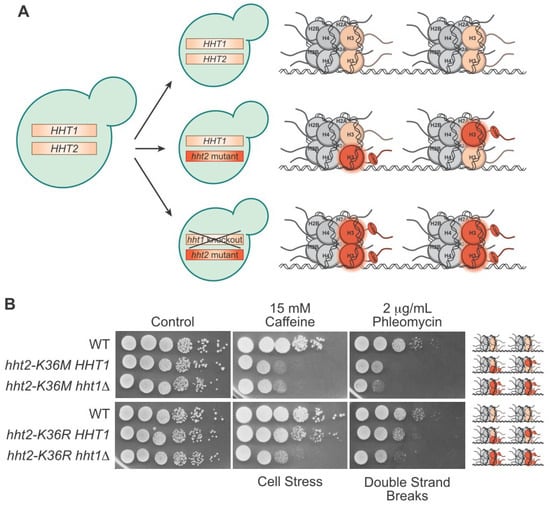
Figure 14A). Studies can then be performed using cells where a single allele is edited and the other allele is wild type, for example
hht2-K36M HHT1, to more closely model what happens in cancer cells where the oncohistone allele is dominant to the numerous wild type alleles of any histone gene (
Figure 14A). Alternatively, if the locus encoding the second histone gene is deleted, this creates a model where the oncohistone variant is the sole histone protein expressed in cells (for example
hht2-K36M hht1Δ) (
Figure 14A). This latter experimental design allows direct analysis of how a specific oncohistone mutant alters histone function. These two scenarios provide the flexibility to assess (1) dominant phenotypes in the presence of the other wildtype histone gene, which aligns with what happens in oncohistone-driven cancers where only a single allele among many is altered and (2) recessive phenotypes in the absence of any wildtype histone gene. The ease of generating these models allows for the rapid screening of a large number of potential oncohistone mutations to assess whether they are likely to alter histone function.
Figure 14. The strength of modeling oncohistone mutants in budding yeast. (A) Budding yeast can be easily engineered to maintain wildtype H3 genes (top), express one mutant and one wildtype H3 (middle), or express only mutant H3 with the wildtype knocked out (bottom). The resulting ratio of wildtype to mutant H3 proteins in a pair of nucleosomes for the given genotypes is depicted on the right. (B) In this serial dilution growth assay, budding yeast cultures were diluted to OD = 5 and serially diluted tenfold before plating onto control or drug plates. The analysis compares H3K36 mutant cells that either have one mutant and one wildtype histone protein (hht2-K36M/R HHT1) and cells that contain mutant histone as the sole copy of histone H3 (hht2-K36M/R hht1Δ) to control wildtype (WT) cells. Cells on control plates were grown for two days, cells on caffeine were grown for five days, and cells on phleomycin were grown for three days. The cellular damage caused by the drugs is indicated below the plates, and the ratio of wildtype to mutant H3 in the nucleosomes within each model is depicted to the right.
Another advantage of the yeast model system is the ability to screen for numerous phenotypes by plating oncohistone models on plates under a variety of conditions, including different temperatures, nutrient sources, and drugs that disrupt different pathways
[18][96] (
Figure 14B). Such growth assays have been exploited extensively to systematically analyze the functional importance of different histone residues for specific PTMs and cell fitness
[19][20][21][22][97,98,99,100]. As an example of this modeling, researchers performed a serial dilution assay of the oncohistone H3K36M and the mutant H3K36R, which has been identified in T-cell acute lymphoblastic leukemia, though its oncogenicity is uncertain
[23][101], and is expressed both in the presence of wildtype H3 (
hht2-K36M/R HHT1 cells) and as the sole copy of H3 (
hht2-K36M/R hht1Δ) (
Figure 14B). To illustrate this experimental approach, results of such an experiment are presented in
Figure 14B. For this experiment, yeast cells were grown in culture, serially diluted, and plated on control plates, plates that contain caffeine, which induces cellular stress
[24][102], or plates that contain phleomycin, which induces double-strand breaks
[25][103]. H3K36M cells show sensitivity to cellular stress and double-strand breaks at fairly similar levels in the presence or absence of wildtype proteins. On the other hand, H3K36R cells show an increase in sensitivity to cellular stress and double-strand breaks when H3K36R is expressed as the sole copy of histone H3 (
Figure 14B). Analyses such as these can provide insight into the genetic and biochemical properties of oncohistone mutants.
Yeast oncohistone models are valuable for extending such an analysis to explore specific amino acid changes identified in cancer patients to identify mutations that alter histone function. For example, a previous investigation assessed the effects of cancer-associated core histone mutants on chromatin remodeling (104). A humanized yeast library was created and employed to discover that certain histone mutants increase histone exchange and nucleosome sliding
[26][104]. Additionally, when these histone mutants were expressed in mammalian cells, it was discovered that cancer-associated gene pathways were upregulated
[26][104]. This investigation is a helpful example of how the power of yeast genetics can be exploited for the rapid discovery and translational studies of human disease-related mutants.
Given the ease of genetic manipulation and unbiased screens in yeast, one might consider expressing a fully humanized nucleosome in the model system. A previous study deleted the core histone genes in
S. cerevisiae and performed a plasmid shuffle to express only the human histone proteins
[27][105]. This process occurred at very low rates, and cells accumulated many suppressor mutations to accommodate growth. Altering two residues in H3 (P121K, Q125K) and three residues in H2A (Q113H, A114Q, V115N) back to the
S. cerevisiae sequence greatly improved efficiency of “humanization”. Interestingly, humanized nucleosomes revealed similar spacing between nucleosomes to that found in wildtype yeast and caused an overall reduction in total RNA production
[27][105]. Although the histone protein sequences are very similar between humans and budding yeast, these differences, in combination with the species–specific epigenetic factors and chromatin remodelers, may be sufficient to cause the phenotypes observed in the humanized strain. Certain experimental questions may be best answered with budding yeast which expresses fully humanized nucleosomes, but the consequences of this expression on chromatin and RNA production would need to be considered.
3. Progress towards Histone and Oncohistone Characterization via Yeast Models
Epigenetic discoveries in yeast have contributed to our understanding of the chromatin-driven regulation of gene expression. Specific examples include modulators of histone acetylation and their role in regulating transcription. The deacetylase Rpd3 (human HDAC3) was originally identified in budding yeast through a screen designed to identify transcriptional regulators, which then enabled the discovery of the related deacetylase in mammalian cells
[28][29][106,107]. The acetyl transferase Gcn5 (human GCN5/KAT2A) was also first discovered and investigated in budding yeast
[30][31][108,109]. Moreover, characterization of the Swi-Independent (SIN) histone mutants helped elucidate the fact that the SWI/SNF complex is critical for chromatin remodeling and revealed histone residues that are involved in DNA–protein interactions
[32][110]. Additionally, the power of yeast genetics is evident in screens that explored the critical roles played by specific histone residues. The results from these screens proved to be foundational to our current understanding of histones. Tandem alanine mutants in histone H3 revealed specific sensitivities to DNA damaging agents and that the αN helix is crucial for its chaperone function, particularly for nucleosome assembly and disassembly
[33][111]. Another study that examined alanine or PTM mimetic mutants of H3 and H4 provided insight into which histone residues influence chronological lifespan
[34][112]. This analysis identified various residues and epigenetic marks that either extended or reduced lifespan which may correlate with destabilizing histone–DNA or histone–histone interactions
[34][112]. An additional investigation discovered surface-accessible residues in all of the core histone proteins that are critical for DNA-interacting functions, such as transcription initiation, transcription elongation, and DNA repair
[22][100]. Furthermore, alanine screening of H4 revealed three adjacent C-terminal residues (L97, Y98, and G99), all of which are conserved in humans, that are required to protect cells from genome instability and ensure proper histone occupancy across the genome
[35][113]. These systematic epigenetic investigations performed in budding yeast helped to provide a foundational understanding of histone protein structure and function, and the resulting discoveries propelled mammalian epigenetic research into an analysis of disease-relevant mutations, such as oncohistones. Thus, there is great power in employing model organisms to explore mutational landscapes, including oncohistone mutants.
Despite the fact that yeast does not develop cancer, taking advantage of these approaches has revealed altered growth patterns associated with yeast oncohistone models. In budding yeast, the expression of either H3K36M or H3K36R as the sole copy of H3 results in reduced growth in the presence of caffeine, suggesting the sensitivity of these oncohistone models to cellular stress
[36][114]. This caffeine sensitivity is also observed when methylation at H3K36 is prevented by deleting the
SET2 gene which encodes the H3K36 methyltransferase
[37][40]. Notably, yeast Set2 also binds to H3K36M, as does human SETD2
[10][90]. As described above, one of the greatest strengths of using yeast as a model system is the ability to perform unbiased screens. To identify factors and pathways upon which H3K36 mutants are specifically sensitive, researchers took advantage of the caffeine-sensitive growth of H3K36 mutant cells in the absence of wild type protein to perform a high copy suppressor screen
[36][114]. The goal of such a screen is to identify suppressors that may identify therapeutically actionable pathways in the corresponding cancers. Researchers identified various suppressors of growth defects on caffeine
[36][114], some of which have known roles in regulating histone function, such as the lysine acetyltransferase Esa1
[38][115], and a putative transcriptional regulator that interacts with Rpd3 and Set3 histone deacetylase complexes, Tos4
[39][116]. Further experimentation to elucidate the mechanism of suppression has the potential to provide insight into mechanisms that underlie altered histone function in cancer.
Many other investigations of oncohistone mutants in yeast have provided insight into the physiological changes that could contribute to oncogenesis. H3G34R mutants in fission yeast result in reduced levels of methylation and acetylation at H3K36
[40][67]. H3G34V mutants in fission yeast reveal sensitivity to induced DNA double-strand breaks, while H3G34R mutants are sensitive to DNA replication stress and defective in homologous recombination
[41][42][78,117]. These results are congruent with what has been found in humans: that cells expressing mutant H3G34 display heightened frequency of genomic mutations
[43][69]. H3K27 methylation is absent in
S. cerevisiae and
S. pombe, possibly because their mechanisms for inhibiting gene transcription are distinct from the heterochromatin present in mammalian systems
[11][91]. Acetylation at this site does occur in yeast, however
[44][118]. Due to this inconsistency with human epigenetic regulation, as well as the absence of the PRC2 complex as described above, yeast has not been employed as a model for the H3K27M oncohistone.
The strengths of employing yeast models to characterize novel oncohistones are also exemplified through investigations of putative oncohistones and characterizing possible oncohistone mechanisms. The variant H2BE76K, which occurs in bladder and head and neck cancers, is found in the globular domain of histone H2B
[45][76]. In budding yeast, engineering one H2B gene to harbor the H2BE76K mutation while maintaining one wildtype copy causes temperature sensitivity and reduced nucleosome stability
[45][76]. Additionally, an array of variants in histone residues located in the acidic patch or histone–DNA interface that are common in cancers displayed increased chromatin remodeling processes and lethal growth defects in fission yeast, suggesting that cancer-related mutants in these residues could also be investigated in the model organism
[26][104]. Finally, as many cancer-associated mutations result in lysine-to-methionine variants (K-to-M), understanding the mechanism by which these changes disrupt cellular growth is crucial. The H3K9M mutant in fission yeast impaired global methylation at H3K9, which is found in constitutive heterochromatin, and displayed enhanced interactions with the human methyltransferase G9a
[46][119]. This mechanism of a K-to-M mutant resulting in tighter association with its respective modifying enzyme is similar to what is observed in the H3K27M and H3K36M oncohistones
[3][4][47][48][61,62,68,70]. Thus, yeast can be employed to model this mode of oncohistone mutant. Yeast model organisms have proven to be useful not only for initial fundamental histone investigations, but also for the ongoing characterization of clinically relevant oncohistone mutants. The genetic simplicity of these models, the ability to readily screen for phenotypes, and the power of genetic screens will continue to provide insight into disease etiology that can be readily extended to humans.