Anti-CD19 chimeric antigen receptor (CAR)-T cell therapy has led to a treatment paradigm shift for B-cell non-Hodgkin lymphomas, first with the approval for relapsed/refractory (R/R) large B-cell lymphomas and subsequently for R/R mantle cell and follicular lymphoma. Many efforts are continuously being made to extend the therapeutic setting in the lymphoma field. Several reports are supporting the safety and efficacy of CAR-T cells in patients with central nervous system disease involvement. Anti-CD30 CAR-T cells for the treatment of Hodgkin lymphoma are in development and early studies looking for the optimal target for T-cell malignancies are ongoing. Anti-CD19/CD20 and CD19/CD22 dual targeting CAR-T cells are under investigation in order to increase anti-lymphoma activity and overcome tumor immune escape. Allogeneic CAR product engineering is on the way, representing a rapidly accessible ‘off-the-shelf’ and potentially more fit product.
- CAR-T cells
- B-cell lymphoma
- central nervous system
1. Introduction
2. Upcoming Settings
2.1. Follicular Lymphoma
Treating follicular lymphoma (FL) patients with anti-CD19 CAR-T cells in clinical practice is already possible, since axi-cel and tisa-cel recently received Food and Drug Administration (FDA) and European Medicine Agency (EMA) approval. In the US, both products are indicated after at least 2 prior lines of therapy, while in Europe, axi-cel requires 3 previous lines [10,11,14,15][10][11][14][15]. Axi-cel was authorized based on the data from the pivotal ZUMA-5 trial. Eligible patients had R/R FL after ≥2 lines of therapy, including an anti-CD20 monoclonal antibody plus an alkylating agent. Among 124 treated patients, 36-month PFS and OS rates were 54.4% and 75%, respectively. Grade ≥ 3 cytokine release syndrome (CRS) occurred in 6% of the patients, and grade ≥ 3 immune effector cell-associated neurotoxicity syndrome (ICANS) occurred in 15% [28,29][27][28]. A propensity score matching comparison between patients with R/R FL treated in the ZUMA-5 setting and patients treated with SOC therapies strongly favored axi-cel both in terms of PFS (HR 0.28, 95% CI: 0.17–0.45) and OS (HR 0.52, 95% CI: 0.28–0.95) [30][29]. Tisa-cel was approved based on the ELARA trial, which also included patients with R/R FL after ≥2 lines of therapy. Ninety-four patients were treated, and the estimated 24-month PFS and OS were 57.4% and 87.7%, respectively. Toxicities were seemingly lower than those of axi-cel, with no CRS exceeding grade 2 and grade ≥ 3 ICANS of 3% [31][30]. Again, a propensity score matching study confirmed the stronger efficacy of CAR-T cell therapy compared to usual care both in terms of PFS and OS [32][31].2.2. Other Indolent Non-Hodgkin Lymphomas
In addition to FL cases, 28 patients with R/R marginal zone lymphoma (MZL) were treated with axi-cel in the ZUMA-5 trial. At a median follow up of 31.8 months, the median PFS was not reached. Grade ≥ 3 CRS occurred in 9% of the patients, while, surprisingly, the grade ≥ 3 ICANS rate was 36%, which was significantly higher than that observed in FL. The reason for this discrepancy is not known, but it could be related to a stronger and more prolonged CAR-T cell expansion in patients with MZL [28,29,35][27][28][32]. Very small reports of CAR-T cell use for Waldenstrom macroglobulinemia (WM)/lymphoplasmacytic lymphoma are available. Three heavily pretreated patients infused with two different anti-CD19 CAR-T cell products attained a clinical response; however, all three had recurrent disease between 3 and 26 months after the initial treatment [36][33]. More recently, a promising report of an anti-CD20 CAR-T product infused in four heavily pretreated and Bruton-tyrosine kinase inhibitor (BTKi) refractory patients has been published: the authors reported one death in complete remission 7 months after treatment due to COVID-19 infection and three patients were free from progression at 1.5–19 months, with manageable CAR-T cell-related toxicities [37][34].3. Extending the Setting for Large B-Cell Lymphoma
3.1. CAR-T Cells for Lymphomas with Central Nervous System Involvement
LBCL with CNS involvement, both isolated (primary CNS lymphoma, PCNSL) or concomitant with systemic disease (secondary CNS lymphoma, SCNSL), represents a rare subset with particularly unfavorable outcomes, especially in cases of disease relapse or frontline refractoriness [70,71][35][36]. The need for agents capable of penetrating the blood–brain barrier (BBB) has led to specific management paradigms for PCNSL and SCNSL, which therefore have been excluded from many LBCL clinical trials with novel agents [72][37]. The adoption of CAR-T cells in this setting was limited by the concern of potential severe neurological toxicities. This fear was supported by the identification of a brain mural pericyte cell population with CD19 antigen expression, representing a potential off-tumor target for anti-CD19 CAR-T cells [73][38]. The capability of peripheral expansion of CAR-T cells, CNS trafficking and subsequent therapeutic activity were first highlighted in cases of ALL and primary brain solid tumors [74,75][39][40]. In 2017, Abramson et al. [76][41] described for the first time a case of LBCL with synchronous systemic and parenchymal brain disease recurrence treated with anti-CD19 CAR-T cells (liso-cel), obtaining a complete response with no CRS or neurotoxicity observed. Afterwards, a rising number of case series confirmed the efficacy and safety of both axi-cel and tisa-cel for R/R LBCL with CNS recurrence, comparable to non-CNS LBCL [77,78,79,80,81][42][43][44][45][46]. In the case series reported by Frigault M et al. [78][43], six of eight patients presented isolated CNS disease at the time of tisa-cel infusion and three of six obtained a clinical response, confirming the potential capability of CAR-T cells to expand even in the absence of systemic disease. Supported by the early evidence of efficacy against SCNSL, CAR-T cells have been tested against R/R PCNSL. In a retrospective analysis of five patients with R/R PCNSL treated with an academic anti-CD19 CAR-T product at City of Hope, three of five patients achieved CR at day 28 post-infusion. Given the rarity of the scenario, the available data for CAR-T cell therapy in patients with CNS lymphoma are so far limited to very small case series. Thus, Cook MR et al. [84][47] conducted a meta-analysis of patients with PCNSL or SCNSL treated with CAR-T cells both in clinical trials and in the real-life setting. One hundred twenty-eight patients treated with axi-cel, tisa-cel, liso-cel or other investigational anti-CD19 CAR-T compounds were included in the analysis (n = 30 with PCNSL, n = 98 with SCNSL) and the authors could confirm the efficacy and tolerability of CAR-T cells in patients with CNS disease. The CRS incidence was superimposable for PCNSL and SCNSL and was observed in around 70% of the patients, with roughly 10% being grade 3–4. ICANS occurred in 50% of the patients and a higher proportion of grade 3–4 cases was reported in SCNSL patients (26%) vs. PCNSL patients (18%), possibly because the presence of a systemic disease might translate into higher systemic inflammation and circulating cytokine levels. Fifty-six percent of PCNSL patients and 47% of SCNSL patients achieved CR as the best response and approximately 37% of the patients in both groups were in ongoing CR 6 months after the infusion. Of note, the authors observed higher toxicities and possibly superior efficacy for CD28 costimulatory domain-based products compared to 4-1BB compounds, similar to LBCL without CNS involvement. Lastly, the German Lymphoma Group just reported the largest retrospective series of SCNSL treated with anti-CD19 CAR-T cells so far, including 28 consecutive patients, half receiving axi-cel and half receiving tisa-cel. Sixty-four percent of the patients responded, with a CR rate of 32% and a 12-month PFS of 40%, which was significantly better for patients treated with axi-cel than tisa-cel (12-month PFS of 62% vs. 19%, respectively). Of note, comparing the 28 SCNSL patients with 168 consecutive patients without CNS involvement treated with CAR-T cells, no differences in terms of survival or toxicity were observed [85][48].3.2. Frontline Adoption of CAR-T Cells
The anticipation of CAR-T cell therapy from the third to second line demonstrated a significant benefit for refractory or early relapsed patients with LBCL in two of the three randomized phase III trials mentioned above, leading to the approval of axi-cel and liso-cel in this setting [19,20,21][19][20][21]. Following these favorable results, CAR-T cells have been explored as first-line therapy for high-risk LBCL patients. Axi-cel has been first tested as part of the frontline treatment in the phase II single arm ZUMA-12 trial [88][49]. In the study, 40 patients with high-risk LBCL, defined by an IPI score ≥ 3 or double/triple-hit histology and with a PET-guided incomplete response after two courses of R-CHOP (based on the Deauville criteria) [89][50], were treated with axi-cel. The trial met the primary end-point, reaching high rates of response (ORR 89%), with a 78% CR rate and 73% of the patients in a continuous response after a median follow-up of almost 16 months. Next, the phase III ZUMA-23 trial has been designed to prove the real benefit of axi-cel over the SOC in the frontline setting [90][51]. Treatment-naïve patients with very high-risk LBCL, defined by an IPI score ≥ 4, are randomized to receive axi-cel or continue with SOC following a first cycle of chemoimmunotherapy (R-CHOP or rituximab, etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin [R-EPOCH]). The trial is currently ongoing and will hopefully demonstrate whether a frontline CAR-T cell approach may be the best option for selected high-risk LBCL patients.4. New Targets for B-NHLs: Multispecific CAR-T Cells
Long-term PFS of patients with aggressive NHL treated with anti-CD19 CAR-T cells remains around 30–40% [91,92][52][53] and about a third of the patients who relapse show CD19 negativity in tumor biopsy samples due to selection of CD19-negative clones and antigen downregulation [24][23]. Thus, multi-targeted CAR-T cell constructs are being developed to overcome this issue [93,94][54][55]. Multiple approaches have been adopted to target more than one tumor antigen. Different single targeting CAR-T cell products can be infused in the same patient at the same time or sequentially. T-cells can also be transduced by different vectors at the same time, creating a population of cells that express different combinations of CARs. A single bicistronic vector encoding two different CARs can be used to create a single T-cell population expressing both CARs. Lastly, a single vector can also be used to encode two CARs on the same receptor (tandem CAR). No data comparing the different approaches is available [95][56]. The different strategies could also be combined in the treatment of the same patient and are represented in Figure 1.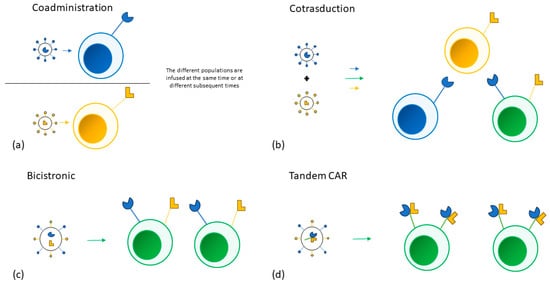
5. New Lymphoma Settings
5.1. Hodgkin Lymphoma
Despite the success of frontline therapy, 20–30% of patients with classical Hodgkin lymphoma (cHL) progress or relapse during the course of their disease history. Salvage with high-dose chemotherapy followed by ASCT remains the standard of care in second-line treatment. Targeted therapy with the anti-CD30 antibody-drug conjugate brentuximab vedotin and immunotherapy with anti PD1/PDL1 are currently considered the best options for subsequent relapses and, more recently, their introduction in combination with chemotherapy regimens in the upfront setting further improved outcomes for patients with advanced stage disease [105,106,107][62][63][64]. Nevertheless, a significant fraction of patients needs alternative approaches. cHL is characterized by a reduced number of malignant Hodgkin and Reed–Sternberg (HRS) cells and an abundance of inflammatory and immune cells, including reactive T- and B-lymphocytes, macrophages, granulocytes and fibroblast-like cells. CD30, a member of the TNF superfamily, is selectively over-expressed in HRS cells with very low expression in normal tissues, and therefore it is considered a promising target for novel treatments.5.2. T-Cell Lymphomas
Despite their rapidly expanding use against B-cell malignancies, the development of CAR-T cell therapy for T-cell neoplasms is rather challenging and no products are currently available for this subset of lymphomas. Most antigens tested as CAR-T cell targets for T-cell neoplasms (such as CD3, CD5, and CD7) are also expressed by normal T-cells [110][65]. Therefore, the lack of a tumor-specific antigen can cause the eradication of normal T-lymphocytes by CAR-T cell treatment, leading to T-cell aplasia, a possibly life-threatening phenomenon [111,112][66][67]. Additionally, the expression of their own target antigens by CAR-T cells leads to mutual killing, also known as fratricide, resulting in impaired persistence and efficacy [113][68]. Although less common than in ALL, circulating tumor cells might be found in peripheral blood. Therefore, T-cell collection could be contaminated with malignant cells, which can be erroneously transduced, as reported in a patient who relapsed by expressing anti-CD19 CAR-tumor cells [114][69]. One of the fundamental challenges in developing effective CAR-T cell therapies for T-cell lymphomas lies in the identification of suitable target antigens. The ideal target should be specific for the malignant cells and not expressed on normal cells, but T-cell lymphomas originate from mature T-cells and lack a clear distinctive cell surface marker.6. ‘Off-the-Shelf’ Products for NHLs: Allogeneic CAR-T and CAR-NK Cells
6.1. Allogeneic CAR-T Cells
In contrast to autologous CAR-T cells, off-the-shelf products offer several potential advantages. First, T-cells are collected from healthy donors, thus avoiding the potential detrimental effects of cancer or and cytotoxic agents. Additionally, large quantities of allo-CAR-Ts can be derived from a single donor, enabling the creation of readily available batches of preserved CAR-T cells for immediate patient access. The increased availability of the product may result in a reduced need for bridging chemotherapy and lower costs [128,129][70][71]. Finally, since allo-CAR-Ts can be created from T-cell subsets that may confer properties such as memory or stemness, a better persistence might be obtained [127,130][72][73]. Despite the potential benefits of allo-CAR-Ts over autologous CAR-T cell products, if immune cells are sourced from MHC-mismatched donors, these products may lead to graft rejection and to the development of graft-versus-host disease (GVHD). Thus, new sources of T-cells for allogeneic approaches have been explored both in preclinical and clinical studies, including virus-specific T-cells, genetically modified conventional T-cells, and non-conventional T-cells [127,131][72][74]. The adoption of virus-specific T-cells represents a promising way for mitigating the GVHD risk, given their established role in treating post-transplant viral infections [132][75]. The safety of this approach was demonstrated in a phase I basket trial involving patients with various B-cell malignancies who received CAR virus-specific T-cells, in which severe GVHD did not occur [133][76]. T-cell genetic modification offers another strategy to address GVHD and rejection concerns by removing endogenous molecules such as αβ T-cell receptors (TCR) and MHC. In an early clinical study, two infants with R/R ALL were successfully treated with universal CAR-T cells, a product in which CD52 and αβ TCR were disrupted through a transcription activator-like effector nuclease (TALEN) gene editing technique [134][77]. While TCR suppression mitigated the GHVD risk, genetic disruption of CD52 expression allowed the adoption of the anti-CD52 alemtuzumab monoclonal antibody as part of the lymphodepletion without affecting CAR-T cell activity. More recently, a CRISPR/Cas9 base-edited anti-CD7 CAR-T product characterized by a triple CD52/CD7/βTCR gene suppression pattern showed efficacy against R/R T-ALL [135][78]. In this case, the gene inactivation of CD52, CD7 and the β chain of the αβ TCR favored the evasion of lymphodepleting therapy, fratricide and GVHD, respectively.6.2. CAR-NK Cells
Natural killer (NK) cells and macrophages are emerging as highly promising candidates for the development of next-generation off-the-shelf CARs, thanks to their advantageous characteristics. NK cells and macrophages are innate immune system components capable of directly recognizing target cells independent of MHC. Importantly, they do not trigger GVHD and their ability to recognize tumor cells even when MHC molecules are downregulated might prevent antigen escape. The efficacy of CAR-NK cells has been demonstrated in preclinical models across a range of hematological and solid tumors [138][79]. Furthermore, in a phase I/II clinical study involving 11 patients with CD19-positive hematological malignancies, promising antitumor effects without significant toxicities were reported following allogeneic UCB-derived CAR-NK cell administration [63][80].6.3. Clinical Experiences with NHLs
The first clinical trial of allo-CAR-Ts (phase I ALPHA study, NCT03939026) showing the safety and feasibility of this approach included patients with R/R LBCL or FL after at least two lines of therapy. These patients received a single infusion of healthy donor-derived CAR-T cells without prior lymphodepletion chemotherapy. The ALLO-501 CAR-T cells are genetically modified anti-CD19 CAR-T cells with disrupted TCR alpha and CD52 genes, thus reducing the risk of GVHD and allowing the use of a humanized anti-CD52 mAb (ALLO-647) for selective and transitory host lymphodepletion. Forty-six out of forty-seven enrolled patients received ALLO-501, and the treatment was initiated rapidly, with a median time of 5 days from enrollment to therapy start. No GVHD was reported and limited ICANS and CRS were observed. Cytopenias occurred in 82.6% of the patients, and grade ≥ 3 infections were observed in 23.9% of the cases, similar to what is observed with autologous CAR-T cells. The 6-month CR rate for LBCL patients was 36.4% [57][81]. The ongoing ALPHA2 study (NCT04416984) is evaluating ALLO-501A (the next-generation product based on ALLO-501 results and lacking the rituximab recognition domains) with either a single or an additional consolidative dose of treatment. In this latter group, patients with ≥SD at day 28 received consolidation with a second ALLO-647 and ALLO-501A cell infusion.7. Other Strategies for CAR-T Cell Manufacturing Improvement
The vector choice for T-cell transduction can also impact CAR activity and clinical outcomes: traditional “always-on” promoters, such as those carried by retroviral vectors, more commonly lead to tonic signaling and overstimulation of CAR-T cells, which can lead to exhaustion and disease relapse. Enhancing immune cell fitness and limiting or interrupting the interaction between CAR-T cells and target antigens are promising strategies to overcome this limitation [142,143,144][82][83][84]. Both these objectives can be reached thanks to the use of next-generation gene modification strategies, such as the use of lentiviral vectors, which do not rely on cell division and have a higher transduction efficiency, or with nonviral methods, such as CRISPR/Cas9 technology. Disruption of the regulation of DNA methylation in CAR-T cells has resulted in enhanced T-cell proliferation and a more prolonged antitumor response, as was first observed in a clinical trial as a result of casual disruption via lentiviral integration of the gene TET2. This observation was later confirmed in a preclinical model by knocking out the gene DNMT3A [145,146][85][86]. The downregulation of the activity of specific genes or the overexpression of target transcription factors can lead to a substantial improvement in T-cell potency, expansion and persistency, and many potential targets are being explored [147][87].8. Conclusions
In the last decade, anti-CD19 CAR-T cells reshaped the treatment paradigm for patients with R/R LBCL and MCL, and many steps forward have been made since the first approval in these settings. Axi-cel and liso-cel have already moved to the second line for refractory and early relapsed patients with LBCL and, more recently, CAR-T cell access has been extended to R/R FL. Several reports demonstrated the efficacy and safety of CAR-T cells in patients with CNS lymphoma and could hopefully support the extension of the regulatory approval to this setting. The use of axi-cel as part of frontline treatment for patients with adverse risk LBCL showed promising results in the phase II single arm ZUMA-12 trial, and the ongoing phase III randomized-controlled ZUMA-23 trial will prove whether this approach will be the new SOC for selected patients with very-high-risk LBCL. New strategies are actively being tested to improve CAR-T cell efficacy and accessibility. Dual targeting CD22/CD19- and CD20/CD19-CAR-T cell products showed significant activity against R/R B-NHLs, emerging as a promising strategy to overcome tumor antigen escape mechanisms. Allogeneic CAR products represent an attractive alternative to autologous CAR-T cells thanks to their ‘off-the-shelf’ nature and potential increased antitumor activity, but further data are necessary to thoroughly assess the long-term implications. Finally, early studies demonstrated promising efficacy of CAR-T cells against new disease subtypes, including R/R cHL and, despite the challenges, T-cell lymphoma.References
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. New Engl. J. Med. 2018, 378, 439–448.
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy Bb2121 in Relapsed or Refractory Multiple Myeloma. New Engl. J. Med. 2019, 380, 1726–1737.
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T-Cell Therapy in Patients with Relapsed or Refractory Multiple Myeloma (CARTITUDE-1): A Phase 1b/2 Open-Label Study. Lancet 2021, 398, 314–324.
- Coscia, M.; Vitale, C.; Cerrano, M.; Maffini, E.; Giaccone, L.; Boccadoro, M.; Bruno, B. Adoptive Immunotherapy with CAR Modified T Cells in Cancer: Current Landscape and Future Perspectives. Front. Biosci.-Landmark 2019, 24, 1284–1315.
- Poletto, S.; Novo, M.; Paruzzo, L.; Frascione, P.M.M.; Vitolo, U. Treatment Strategies for Patients with Diffuse Large B-Cell Lymphoma. Cancer Treat Rev. 2022, 110, 102443.
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. New Engl. J. Med. 2017, 377, 2531–2544.
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. New Engl. J. Med. 2019, 380, 45–56.
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene Maraleucel for Patients with Relapsed or Refractory Large B-Cell Lymphomas (TRANSCEND NHL 001): A Multicentre Seamless Design Study. Lancet 2020, 396, 839–852.
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-Term Safety and Activity of Axicabtagene Ciloleucel in Refractory Large B-Cell Lymphoma (ZUMA-1): A Single-Arm, Multicentre, Phase 1-2 Trial. Lancet Oncol. 2019, 20, 31–42.
- YESCARTA (Axicabtagene Ciloleucel)|FDA. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/yescarta-axicabtagene-ciloleucel (accessed on 10 October 2023).
- Yescarta | European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/yescarta (accessed on 10 October 2023).
- BREYANZI (Lisocabtagene Maraleucel)|FDA. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/breyanzi-lisocabtagene-maraleucel (accessed on 10 October 2023).
- Breyanzi | European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/breyanzi (accessed on 10 October 2023).
- KYMRIAH (Tisagenlecleucel)|FDA. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/kymriah-tisagenlecleucel (accessed on 10 October 2023).
- Kymriah | European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/kymriah (accessed on 10 October 2023).
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. New Engl. J. Med. 2020, 382, 1331–1342.
- TECARTUS (Brexucabtagene Autoleucel)|FDA. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/tecartus-brexucabtagene-autoleucel (accessed on 10 October 2023).
- Tecartus|European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/tecartus (accessed on 10 October 2023).
- Locke, F.L.; Miklos, D.B.; Jacobson, C.A.; Perales, M.-A.; Kersten, M.-J.; Oluwole, O.O.; Ghobadi, A.; Rapoport, A.P.; McGuirk, J.; Pagel, J.M.; et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. New Engl. J. Med. 2022, 386, 640–654.
- Kamdar, M.; Solomon, S.R.; Arnason, J.; Johnston, P.B.; Glass, B.; Bachanova, V.; Ibrahimi, S.; Mielke, S.; Mutsaers, P.; Hernandez-Ilizaliturri, F.; et al. Lisocabtagene Maraleucel versus Standard of Care with Salvage Chemotherapy Followed by Autologous Stem Cell Transplantation as Second-Line Treatment in Patients with Relapsed or Refractory Large B-Cell Lymphoma (TRANSFORM): Results from an Interim Analysis of an Open-Label, Randomised, Phase 3 Trial. Lancet 2022, 399, 2294–2308.
- Bishop, M.R.; Dickinson, M.; Purtill, D.; Barba, P.; Santoro, A.; Hamad, N.; Kato, K.; Sureda, A.; Greil, R.; Thieblemont, C.; et al. Second-Line Tisagenlecleucel or Standard Care in Aggressive B-Cell Lymphoma. New Engl. J. Med. 2022, 386, 629–639.
- Cerrano, M.; Ruella, M.; Perales, M.A.; Vitale, C.; Faraci, D.G.; Giaccone, L.; Coscia, M.; Maloy, M.; Sanchez-Escamilla, M.; Elsabah, H.; et al. The Advent of CAR T-Cell Therapy for Lymphoproliferative Neoplasms: Integrating Research into Clinical Practice. Front. Immunol. 2020, 11, 888.
- Plaks, V.; Rossi, J.M.; Chou, J.; Wang, L.; Poddar, S.; Han, G.; Wang, Z.; Kuang, S.Q.; Chu, F.; Davis, R.E.; et al. CD19 Target Evasion as a Mechanism of Relapse in Large B-Cell Lymphoma Treated with Axicabtagene Ciloleucel. Blood 2021, 138, 1081–1085.
- Jain, M.D.; Zhao, H.; Wang, X.; Atkins, R.; Menges, M.; Reid, K.; Spitler, K.; Faramand, R.; Bachmeier, C.; Dean, E.A.; et al. Tumor Interferon Signaling and Suppressive Myeloid Cells Are Associated with CAR T-Cell Failure in Large B-Cell Lymphoma. Blood 2021, 137, 2621–2633.
- Allen, E.S.; Stroncek, D.F.; Ren, J.; Eder, A.F.; West, K.A.; Fry, T.J.; Lee, D.W.; Mackall, C.L.; Conry-Cantilena, C. Autologous Lymphapheresis for the Production of Chimeric Antigen Receptor T Cells. Transfusion 2017, 57, 1133–1141.
- Jo, T.; Yoshihara, S.; Okuyama, Y.; Fujii, K.; Henzan, T.; Kahata, K.; Yamazaki, R.; Takeda, W.; Umezawa, Y.; Fukushima, K.; et al. Risk Factors for CAR-T Cell Manufacturing Failure among DLBCL Patients: A Nationwide Survey in Japan. Br. J. Haematol. 2023, 202, 256–266.
- Jacobson, C.; Chavez, J.C.; Sehgal, A.R.; William, B.M.; Munoz, J.; Salles, G.; Munshi, P.N.; Casulo, C.; Maloney, D.; de Vos, S.; et al. Primary Analysis of Zuma-5: A Phase 2 Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients with Relapsed/Refractory (R/R) Indolent Non-Hodgkin Lymphoma (INHL). Blood 2020, 136, 40–41.
- Neelapu, S.S.; Chavez, J.; Sehgal, A.R.; Epperla, N.; Ulrickson, M.; Bachy, E.; Munshi, P.N.; Casulo, C.; Maloney, D.G.; de Vos, S.; et al. 3-Year Follow-up Analysis of ZUMA-5: A Phase 2 Study of Axicabtagene Ciloleucel (Axi-Cel) in Patients with Relapsed/Refractory (R/R) Indolent Non-Hodgkin Lymphoma (INHL). Blood 2022, 140, 10380–10383.
- Palomba, M.L.; Ghione, P.; Patel, A.R.; Nahas, M.; Beygi, S.; Hatswell, A.J.; Kanters, S.; Limbrick-Oldfield, E.H.; Wade, S.W.; Ray, M.D.; et al. A 24-Month Updated Analysis of the Comparative Effectiveness of ZUMA-5 (Axi-Cel) vs. SCHOLAR-5 External Control in Relapsed/Refractory Follicular Lymphoma. Expert Rev. Anticancer Ther. 2023, 23, 199–206.
- Dreyling, M.; Dickinson, M.; Martinez Lopez, J.; Kolstad, A.; Butler, J.P.; Ghosh, M.; Popplewell, L.L.; Chavez, J.; Bachy, E.; Kato, K.; et al. Long-Term Clinical Outcomes and Correlative Efficacy Analyses in Patients (Pts) with Relapsed/Refractory Follicular Lymphoma (r/r FL) Treated with Tisagenlecleucel in the Elara Trial. Blood 2022, 140, 1459–1463.
- Salles, G.; Schuster, S.J.; Dreyling, M.; Fischer, L.; Kuruvilla, J.; Patten, P.E.M.; von Tresckow, B.; Smith, S.M.; Jiménez-Ubieto, A.; Davis, K.L.; et al. Efficacy Comparison of Tisagenlecleucel vs Usual Care in Patients with Relapsed or Refractory Follicular Lymphoma. Blood Adv. 2022, 6, 5835–5843.
- Holtzman, N.G.; Shah, N.N. CAR T-Cell Therapy for Indolent Lymphoma: A New Treatment Paradigm? Lancet Oncol. 2022, 23, 6–8.
- Lia Palomba, M.; Qualls, D.; Monette, S.; Sethi, S.; Dogan, A.; Roshal, M.; Senechal, B.; Wang, X.; Rivière, I.; Sadelain, M.; et al. CD19-Directed Chimeric Antigen Receptor T Cell Therapy in Waldenström Macroglobulinemia: A Preclinical Model and Initial Clinical Experience. J. Immunother Cancer 2022, 10, e004128.
- Shadman, M.; Yeung, C.; Redman, M.W.; Lee, S.Y.; Lee, D.H.; Ra, S.; Qian, D.H.; Dezube, B.; Chapuis, A.; Green, D.; et al. P1097: CD20 CAR-T THERAPY WITH MB-106 FOR BTK INHIBITOR-REFRACTORY WALDENSTRÖM MACROGLOBULINEMIA (WM)/LYMPHOPLASMACYTIC LYMPHOMA (LPL)—SINGLE INSTITUTION STUDY. Hemasphere 2023, 7, e68877ca.
- Jahnke, K.; Thiel, E.; Martus, P.; Herrlinger, U.; Weller, M.; Fischer, L.; Korfel, A. Relapse of Primary Central Nervous System Lymphoma: Clinical Features, Outcome and Prognostic Factors. J. Neurooncol. 2006, 80, 159–165.
- Zinzani, P.L.; Magagnoli, M.; Frezza, G.; Prologo, G.; Gherlinzoni, F.; Bendandi, M.; Albertini, P.; Babini, L.; D’Alessandro, R.; Tura, S. Isolated Central Nervous System Relapse in Aggressive Non-Hodgkin’s Lymphoma: The Bologna Experience. Leuk. Lymphoma 1999, 32, 571–576.
- Chihara, D.; Dunleavy, K. Primary Central Nervous System Lymphoma: Evolving Biologic Insights and Recent Therapeutic Advances. Clin. Lymphoma Myeloma Leuk. 2021, 21, 73–79.
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142.e17.
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. New Engl. J. Med. 2013, 368, 1509–1518.
- Keu, K.V.; Witney, T.H.; Yaghoubi, S.; Rosenberg, J.; Kurien, A.; Magnusson, R.; Williams, J.; Habte, F.; Wagner, J.R.; Forman, S.; et al. Reporter Gene Imaging of Targeted T Cell Immunotherapy in Recurrent Glioma. Sci. Transl. Med. 2017, 9, eaag2196.
- Abramson, J.S.; McGree, B.; Noyes, S.; Plummer, S.; Wong, C.; Chen, Y.-B.; Palmer, E.; Albertson, T.; Ferry, J.A.; Arrillaga-Romany, I.C. Anti-CD19 CAR T Cells in CNS Diffuse Large-B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 783–784.
- Novo, M.; Ruff, M.W.; Skrabek, P.J.; Lin, Y. Axicabtagene Ciloleucel Chimeric Antigen Receptor T Cell Therapy in Lymphoma with Secondary Central Nervous System Involvement. Mayo Clin. Proc. 2019, 94, 2361–2364.
- Frigault, M.J.; Maus, M.V.; Dietrich, J.; Martinez-Lage, M.; Leick, M.; Choi, B.D.; DeFilipp, Z.; Chen, Y.-B.; Abramson, J.; Crombie, J.; et al. Tisagenlecleucel CAR T-Cell Therapy in Secondary CNS Lymphoma. Blood 2019, 134, 860–866.
- Ghafouri, S.; Timmerman, J.; Larson, S.; Mead, M.D. Axicabtagene Ciloleucel CAR T-Cell Therapy for Relapsed/Refractory Secondary CNS Non-Hodgkin Lymphoma: Comparable Outcomes and Toxicities, but Shorter Remissions May Warrant Alternative Consolidative Strategies? Bone Marrow Transpl. 2021, 56, 974–977.
- Ahmed, G.; Hamadani, M.; Shah, N.N. CAR T-Cell Therapy for Secondary CNS DLBCL. Blood Adv. 2021, 5, 5626–5630.
- Bennani, N.N.; Maurer, M.J.; Nastoupil, L.J.; Jain, M.D.; Chavez, J.C.; Cashen, A.F.; Dahiya, S.; Lekakis, L.J.; Reagan, P.M.; Oluwole, O.O.; et al. Experience with Axicabtagene Ciloleucel (Axi-Cel) in Patients with Secondary CNS Involvement: Results from the US Lymphoma CAR T Consortium. Blood 2019, 134, 763.
- Cook, M.R.; Dorris, C.S.; Makambi, K.H.; Luo, Y.; Munshi, P.N.; Donato, M.; Rowley, S.; Saad, A.; Goy, A.; Dunleavy, K.; et al. Toxicity and Efficacy of CAR T-Cell Therapy in Primary and Secondary CNS Lymphoma: A Meta-Analysis of 128 Patients. Blood Adv. 2023, 7, 32–39.
- Ayuk, F.; Gagelmann, N.; von Tresckow, B.; Wulf, G.; Rejeski, K.; Stelljes, M.; Penack, O.; Baldus, C.D.; Kröger, N.; Bethge, W.; et al. Real-World Results of CAR T-Cell Therapy for Large B-Cell Lymphoma with CNS Involvement: A GLA/DRST Study. Blood Adv. 2023, 7, 5316–5319.
- Neelapu, S.S.; Dickinson, M.; Munoz, J.; Ulrickson, M.L.; Thieblemont, C.; Oluwole, O.O.; Herrera, A.F.; Ujjani, C.S.; Lin, Y.; Riedell, P.A.; et al. Axicabtagene Ciloleucel as First-Line Therapy in High-Risk Large B-Cell Lymphoma: The Phase 2 ZUMA-12 Trial. Nat. Med. 2022, 28, 735–742.
- Barrington, S.F.; Mikhaeel, N.G.; Kostakoglu, L.; Meignan, M.; Hutchings, M.; Müeller, S.P.; Schwartz, L.H.; Zucca, E.; Fisher, R.I.; Trotman, J.; et al. Role of Imaging in the Staging and Response Assessment of Lymphoma: Consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J. Clin. Oncol. 2014, 32, 3048–3058.
- Nastoupil, L.J. Will CAR T-Cell Therapy Be the Preferred Modality in Frontline Treatment of Large B-Cell Lymphoma? Hematologist 2023, 20, doi.
- Westin, J.R.; Kersten, M.J.; Salles, G.; Abramson, J.S.; Schuster, S.J.; Locke, F.L.; Andreadis, C. Efficacy and Safety of CD19-Directed CAR-T Cell Therapies in Patients with Relapsed/Refractory Aggressive B-Cell Lymphomas: Observations from the JULIET, ZUMA-1, and TRANSCEND Trials. Am. J. Hematol. 2021, 96, 1295–1312.
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554.
- Zah, E.; Lin, M.Y.; Anne, S.B.; Jensen, M.C.; Chen, Y.Y. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol. Res. 2016, 4, 498–508.
- Furqan, F.; Shah, N.N. Multispecific CAR T Cells Deprive Lymphomas of Escape via Antigen Loss. Annu. Rev. Med. 2023, 74, 279–291.
- Shah, N.N.; Maatman, T.; Hari, P.; Johnson, B. Multi Targeted CAR-T Cell Therapies for B-Cell Malignancies. Front. Oncol. 2019, 9, 146.
- Pavlasova, G.; Mraz, M. The Regulation and Function of CD20: An “Enigma” of B-Cell Biology and Targeted Therapy. Haematologica 2020, 105, 1494–1506.
- Zhang, Y.; Wang, Y.; Liu, Y.; Tong, C.; Wang, C.; Guo, Y.; Ti, D.; Yang, Q.; Qiao, S.; Wu, Z.; et al. Long-Term Activity of Tandem CD19/CD20 CAR Therapy in Refractory/Relapsed B-Cell Lymphoma: A Single-Arm, Phase 1-2 Trial. Leukemia 2022, 36, 189–196.
- Meng, Y.; Deng, B.; Rong, L.; Li, C.; Song, W.; Ling, Z.; Xu, J.; Duan, J.; Wang, Z.; Chang, A.H.; et al. Short-Interval Sequential CAR-T Cell Infusion May Enhance Prior CAR-T Cell Expansion to Augment Anti-Lymphoma Response in B-NHL. Front. Oncol. 2021, 11, 640166.
- Schneider, D.; Xiong, Y.; Wu, D.; Hu, P.; Alabanza, L.; Steimle, B.; Mahmud, H.; Anthony-Gonda, K.; Krueger, W.; Zhu, Z.; et al. Trispecific CD19-CD20-CD22-Targeting DuoCAR-T Cells Eliminate Antigen-Heterogeneous B Cell Tumors in Preclinical Models. Sci. Transl. Med. 2021, 13, eabc6401.
- Roddie, C.; Lekakis, L.J.; Marzolini, M.A.V.; Ramakrishnan, A.; Zhang, Y.; Hu, Y.; Peddareddigari, V.G.R.; Khokhar, N.Z.; Chen, R.W.; Basilico, S.; et al. Dual Targeting of CD19 and CD22 with Bicistronic CAR-T Cells in Patients with Relapsed/Refractory Large B-Cell Lymphoma. Blood 2023, 141, 2470–2482.
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illés, Á.; Picardi, M.; et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 378, 331–344.
- Ansell, S.M.; Radford, J.; Connors, J.M.; Długosz-Danecka, M.; Kim, W.-S.; Gallamini, A.; Ramchandren, R.; Friedberg, J.W.; Advani, R.; Hutchings, M.; et al. Overall Survival with Brentuximab Vedotin in Stage III or IV Hodgkin’s Lymphoma. N. Engl. J. Med. 2022, 387, 310–320.
- Herrera, A.F.; LeBlanc, M.L.; Castellino, S.M.; Li, H.; Rutherford, S.C.; Evens, A.M.; Davison, K.; Punnett, A.; Hodgson, D.C.; Parsons, S.K.; et al. SWOG S1826, a Randomized Study of Nivolumab(N)-AVD versus Brentuximab Vedotin(BV)-AVD in Advanced Stage (AS) Classic Hodgkin Lymphoma (HL). J. Clin. Oncol. 2023, 41, LBA4.
- Safarzadeh Kozani, P.; Safarzadeh Kozani, P.; Rahbarizadeh, F. CAR-T Cell Therapy in T-Cell Malignancies: Is Success a Low-Hanging Fruit? Stem Cell Res. Ther. 2021, 12, 527.
- Leonard, W.J. Cytokines and Immunodeficiency Diseases. Nat. Rev. Immunol. 2001, 1, 200–208.
- Alcantara, M.; Tesio, M.; June, C.H.; Houot, R. CAR T-Cells for T-Cell Malignancies: Challenges in Distinguishing between Therapeutic, Normal, and Neoplastic T-Cells. Leukemia 2018, 32, 2307–2315.
- Cooper, M.L.; Choi, J.; Staser, K.; Ritchey, J.K.; Devenport, J.M.; Eckardt, K.; Rettig, M.P.; Wang, B.; Eissenberg, L.G.; Ghobadi, A.; et al. An “off-the-Shelf” Fratricide-Resistant CAR-T for the Treatment of T Cell Hematologic Malignancies. Leukemia 2018, 32, 1970–1983.
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of Resistance to Chimeric Antigen Receptor T Cell Therapy by Transduction of a Single Leukemic B Cell. Nat. Med. 2018, 24, 1499–1503.
- Jeyakumar, N.; Smith, M. Custom CARs: Leveraging the Adaptability of Allogeneic CAR Therapies to Address Current Challenges in Relapsed/Refractory DLBCL. Front. Immunol. 2022, 13, 887866.
- Brudno, J.N.; Somerville, R.P.T.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J. Clin. Oncol. 2016, 34, 1112–1121.
- Chen, Y.J.; Abila, B.; Mostafa Kamel, Y. CAR-T: What Is Next? Cancers 2023, 15, 663.
- Sheikh, S.; Migliorini, D.; Lang, N. CAR T-Based Therapies in Lymphoma: A Review of Current Practice and Perspectives. Biomedicines 2022, 10, 1960.
- Khurana, A.; Lin, Y. Allogeneic Chimeric Antigen Receptor Therapy in Lymphoma. Curr. Treat Options Oncol. 2022, 23, 171–187.
- Leen, A.M.; Bollard, C.M.; Mendizabal, A.M.; Shpall, E.J.; Szabolcs, P.; Antin, J.H.; Kapoor, N.; Pai, S.Y.; Rowley, S.D.; Kebriaei, P.; et al. Multicenter Study of Banked Third-Party Virus-Specific T Cells to Treat Severe Viral Infections after Hematopoietic Stem Cell Transplantation. Blood 2013, 121, 5113–5123.
- Cruz, C.R.Y.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of Donor-Derived CD19-Redirected Virus-Specific T Cells for B-Cell Malignancies Relapsed after Allogeneic Stem Cell Transplant: A Phase 1 Study. Blood 2013, 122, 2956–2973.
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular Remission of Infant B-ALL after Infusion of Universal TALEN Gene-Edited CAR T Cells. Sci. Transl. Med. 2017, 9, eaaj2013.
- Chiesa, R.; Georgiadis, C.; Syed, F.; Zhan, H.; Etuk, A.; Gkazi, S.A.; Preece, R.; Ottaviano, G.; Braybrook, T.; Chu, J.; et al. Base-Edited CAR7 T Cells for Relapsed T-Cell Acute Lymphoblastic Leukemia. New Engl. J. Med. 2023, 389, 899–910.
- Mehta, R.S.; Rezvani, K. Chimeric Antigen Receptor Expressing Natural Killer Cells for the Immunotherapy of Cancer. Front. Immunol. 2018, 9, 283.
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553.
- Neelapu, S.S.; Nath, R.; Munoz, J.; Tees, M.; Miklos, D.B.; Frank, M.J.; Malik, S.A.; Stevens, D.; Shin, C.R.; Balakumaran, A.; et al. ALPHA Study: ALLO-501 Produced Deep and Durable Responses in Patients with Relapsed/Refractory Non-Hodgkin’s Lymphoma Comparable to Autologous CAR T. Blood 2021, 138, 3878.
- Gumber, D.; Wang, L.D. Improving CAR-T Immunotherapy: Overcoming the Challenges of T Cell Exhaustion. EBioMedicine 2022, 77, 103941.
- Irving, M.; Lanitis, E.; Migliorini, D.; Ivics, Z.; Guedan, S. Choosing the Right Tool for Genetic Engineering: Clinical Lessons from Chimeric Antigen Receptor-T Cells. Hum. Gene Ther. 2021, 32, 1044–1058.
- Reichenbach, P.; Giordano Attianese, G.M.P.; Ouchen, K.; Cribioli, E.; Triboulet, M.; Ash, S.; Saillard, M.; Vuillefroy de Silly, R.; Coukos, G.; Irving, M. A Lentiviral Vector for the Production of T Cells with an Inducible Transgene and a Constitutively Expressed Tumour-Targeting Receptor. Nat. Biomed. Eng. 2023, 7, 1063–1080.
- Prinzing, B.; Zebley, C.C.; Petersen, C.T.; Fan, Y.; Anido, A.A.; Yi, Z.; Nguyen, P.; Houke, H.; Bell, M.; Haydar, D.; et al. Deleting DNMT3A in CAR T Cells Prevents Exhaustion and Enhances Antitumor Activity. Sci. Transl. Med. 2021, 13, eabh0272.
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 Promotes the Therapeutic Efficacy of CD19-Targeted T Cells. Nature 2018, 558, 307–312.
- Labanieh, L.; Mackall, C.L. CAR Immune Cells: Design Principles, Resistance and the next Generation. Nature 2023, 614, 635–648.