Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ana Cristina Simões e Silva and Version 2 by Fanny Huang.
Nephrotic syndrome (NS) is a clinical entity characterized by the triad of proteinuria, hypoalbuminemia, and edema. The main pathophysiological alteration in NS is the impairment of the glomerular filtration barrier. Therefore, its permeability becomes non-selective, and urinary protein loss occurs. Being the final barrier to protein loss in the glomerulus, alterations in podocyte structure can explain why injuries in these cells, such as the effacement of their foot processes, are typically related to proteinuria and hypoalbuminemia and, therefore, to the conditions under the NS umbrella.
- nephrotic syndrome
- podocyte
- proteinuria
1. Introduction
Nephrotic syndrome (NS) is a clinical entity characterized by the triad of proteinuria, hypoalbuminemia, and edema [1]. It is a group of diseases composed of numerous etiologies, ranging from congenital, such as Finnish Type Nephropathy and Diffuse Mesangial Sclerosis, to primary renal diseases, including Minimal Change Nephrotic Syndrome (MCNS) and Focal Segmental Glomerulosclerosis (FSGS), or secondary forms due to systemic diseases such as, for instance, diabetes mellitus and lupus erythematosus [2]. The reported incidence of childhood NS is 2–7 per 100,000 children, with a prevalence of about 16 cases per 100,000 and variability among ethnic groups. The peak age of onset of childhood NS occurs at 2–3 years in most cases [3]. MCNS is the most common histological variant, accounting for approximately 80% of NS cases in children, followed by FSGS, which, although less common, is more related to poor long-term outcomes [4][5][4,5].
The main pathophysiological alteration in NS is the impairment of the glomerular filtration barrier. Therefore, its permeability becomes non-selective, and urinary protein loss occurs. All components of the glomerular barrier may interfere with the permeability of molecules. These components include the fenestrated endothelium, the glomerular basement membrane (GBM), and the podocytes. In NS patients, the effacement of the foot processes of the podocytes is a common finding in the electronic microscopy of the kidney tissue [6]. Podocytes are highly differentiated epithelial cells with a large cell body and elongated cellular extensions, the foot processes, that interdigitate along the outer wall of the glomerular capillary and, among other functions, provide structural support and control the filtration process [7].
Being the final barrier to protein loss in the glomerulus, alterations in podocyte structure can explain why injuries in these cells, such as the effacement of their foot processes, are typically related to proteinuria and hypoalbuminemia and, therefore, to the conditions under the NS umbrella [6].
2. Pathophysiological Mechanisms of the Podocytopathies
2.1. Pathophysiology of Nephrotic Syndrome and the Glomerular Filtration
Despite often occurring rapidly, NS can have an insidious presentation, with proteinuria before the onset of symptoms. The proteinuria indicates an altered permeability of the glomerular filtration barrier [8] and contributes to edema and hypoalbuminemia. Hypoalbuminemia induces lipoprotein synthesis, which causes hyperlipidemia [2][3][2,3]. The three components of the glomerular filtration barrier—the fenestrated endothelium, the GBM, and the podocytes—control the size of filtered molecules [9]. The podocytes are visceral glomerular epithelial cells, and their injury plays an important role in the development of the NS. Therefore, the main pathophysiological alterations in NS justify the view of this syndrome as a podocytopathy [2].
Podocytes are highly specialized and polarized epithelial cells related to the synthesis of extracellular matrix components of the GBM. Furthermore, these cells have a structural role in supporting the glomerular capillaries with their interdigitating foot processes (FP), which are linked to each other through a special cell–cell junction, the slit diaphragm (SD). The podocyte also synthesizes many proteins from the SD. The unique zipper-like architecture of the SD is essential to the glomerulus permeability; therefore, podocyte depletion due to detachment, apoptosis, or necrosis is a critical step in the development of NS [7].
Electron microscopy shows the effacement of the foot processes as a major factor related to the glomerular lesion [7]. The podocytes and the SD zipper-like array are composed of an extensive group of molecules, including an actin cytoskeleton and proteins such as ZO-1, nephrin, podocin, and CD2AP, in both the podocyte itself and the GBM. Some etiologies of congenital and steroid-resistant NS can be linked to mutations in genes encoding some of these components, altering the glomerular filtration barrier and leading to intense urinary protein loss [1].
The mechanisms that underlie FP effacement and podocytes’ response to injury are important to determine the clinical manifestations of NS and prognosis [6]. Furthermore, the understanding of podocyte-specific protein alterations and the dynamic changes of glomerular permeability and selectivity are also relevant for the management and treatment of NS [2][3][2,3].
2.2. Podocyte Foot Processes: Update on Molecular Anatomy and Effacement
Each podocyte FP contains a contractile system composed of actin, myosin-II, actinin-4, talin, and vinculin [10]. The cytoskeleton molecular composition in podocyte FP is critical to podocyte function and effacement processes. The foot processes (FPs) are anchored to the GBM because of 31 integrin complexes, which, consisting of heterodimeric transmembrane receptors, mediate cell attachment to the extracellular matrix and are crucial for cell signaling. The adjacent FPs of the same podocyte are bundled using actin filaments, which form arches between these adjacent FPs. The connection between neighboring FPs is the SD [6].
The SD is the main size-selective filter barrier in the kidney. The protein composition of SD justifies the importance of this cellular junction for glomerulus permeability. The main proteins of the SD are nephrin, P-cadherin, CD2AP, ZO-1, FAT, podocin, and Neph1 (Figure 1). These contractile proteins allow the maintenance of glomerular filtration and also indicate that the fusion of podocyte FPs and the obliteration of SD are highly related to their alterations, leading to NS [6][11][6,11].
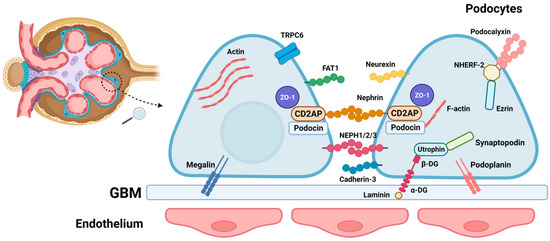
Figure 1. Schematic representation of the glomerular slit diaphragm, a pivotal structure within the glomerulus that is essential for the filtration barrier’s integrity. Positioned between podocyte foot processes and the glomerular basement membrane (GBM), this intricate assembly involves a network of molecules crucial for maintaining structural and functional integrity. Key components include transmembrane proteins like nephrin (a structural protein forming the backbone of the slit diaphragm), podocin (interacting with nephrin, crucial for slit diaphragm function), NEPH1/2/3 (members of the neph family proteins contributing to slit diaphragm molecular architecture), Cadherin 3 (involved in cell adhesion within podocytes), and podoplanin (a transmembrane protein expressed in podocytes). Additionally, molecules such as laminin (a component of the GBM providing structural support) and dystroglycan complex proteins (α-DG, β-DG) (linking the cytoskeleton to the extracellular matrix) are integral to the GBM. The schema also highlights other essential players, including utrophin (participating in cytoskeletal organization), synaptopodin (involved in actin cytoskeleton regulation), ezrin (linking actin filaments to the plasma membrane), NHERF-2 (a scaffold protein interacting with nephrin and podocin), podocalyxin (a sialoprotein contributing to the glycocalyx), FAT1 (a cell adhesion and signaling protein), ZO-1 (a tight junction-associated protein), TRCP6 (an ion channel implicated in slit diaphragm function), actin (a cytoskeletal component crucial for podocyte structure), and megalin (a receptor involved in protein reabsorption in the proximal tubule).
There is another important protein associated with actin microfilaments in FPs, synaptopodin [12], and its expression can indicate alterations in podocytes and also in the response to therapy in many forms of NS [13]. Therefore, synaptopodin likely plays an important role in the podocyte FP molecular composition, interacting with membrane-associated guanylate kinase, WW, and PDZ domain-containing protein 1 (MAGI-1) such as actinin-4 [14]. This may indicate an association between its expression and alterations in the podocyte.
As mentioned, the molecular composition of the cytoskeleton in podocyte FPs is dynamic and determines the maintenance of glomerular filtration. This has been demonstrated by using the acute protamine sulfate (PS)/heparin perfusion model, which dramatically reduces FPs in rodents [15][16][15,16]. Interference with one of the three membrane domains of the basolateral portion of FPs, including the apical membrane domain, the SD complex, and the basal membrane domain, can lead to alterations in the actin cytoskeleton. The consequences of these alterations are the fusion and effacement of podocyte FPs and the obliteration of the SD.
2.3. Histopathology of Common Types of Nephrotic Syndrome in Childhood
The types and causes of NS in childhood vary according to age. The most frequent type of NS in pediatric patients is primary or idiopathic disease, in which the etiology is not defined. When the NS starts before 1 year of age, the possibility of congenital NS should be considered, mostly in cases that occur before 6 months of age. Congenital NS is related to mutations in proteins related to SD. When nephrotic syndrome first appears during the school-age or adolescent years, it is important to consider the possibility of secondary NS due to systemic diseases [17]. The two most common histological forms of primary NS are MCNS and FSGS (Figure 2). Understanding histopathology is important to guide correct molecular and morphologic diagnoses and posterior management of NS [18].
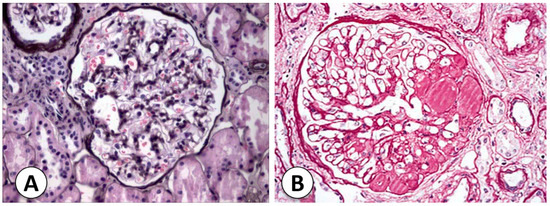
Figure 2. Histological comparison of Minimal Change Nephrotic Syndrome (MCNS) and Focal Segmental Glomerulosclerosis (FSGS). (A) Representative histological slide of kidney tissue from a patient diagnosed with Minimal Change Nephrotic Syndrome (MCNS). The glomerulus exhibits minimal structural alterations, characterized by a lack of significant changes in the glomerular basement membrane, mesangium, and podocytes (Jones silver stain). Electronic microscopy can reveal diffuse effacement of podocyte foot processes, a hallmark feature of MCNS. (B) In contrast, kidney tissue from a patient diagnosed with Focal Segmental Glomerulosclerosis (FSGS) is depicted in this section (h&E staining). The histological examination reveals focal and segmental areas of sclerosis within the glomerulus, indicating regions of glomerular injury and scarring. The presence of hyalinosis, adhesions, and segmental obliteration of capillary lumina is evident, reflecting the characteristic histopathological changes associated with FSGS. Both micrographs were captured at a magnification of 400× (personal archive of the authors).
2.3.1. Congenital Nephrotic Syndrome (Finnish Type)
This form of NS is highly related to prematurity in neonatal and prenatal presentations as well. In Finland, the incidence reaches 1:8200 births and is an autosomal-recessive disorder with heavy proteinuria in the neonatal period. The Finnish nephropathy gene is located on the long arm of chromosome 19 (19q13.1), which codes for nephrin [2]. The Finnish nephropathy gene is located on the long arm of chromosome 19 (19q13.1), which codes for nephrin. Around 70 changes to nephrin have been found in different parts of the body. These changes can be single nucleotide missense mutations, splices, insertions, deletions, or nonsense. Some mutations, such as Fin-major (deletion in exon 2) and Fin-minor (nonsense mutation in exon 26), are the most common. The kidney histopathology may help with congenital NS diagnosis, especially by using electron microscopy and immunohistochemistry [18]. A renal biopsy shows many glomeruli with mesangial hypercellularity and hyperlobulated capillary tufts. Furthermore, proximal and distal tubules may present microcystic dilatation, and the podocytes present FP effacement and/or villous transformations under electron microscopy. Immunohistochemical staining for nephrin is an important method to differentiate FN from other causes of congenital NS [2].
2.3.2. Diffuse Mesangial Sclerosis
The presentation of diffuse mesangial sclerosis (DMS) is similar to that of FN. However, its presentation can be later, up to 4 years of age, but usually persistent NS is described within the first 9 months of life. DMS is the second-most common cause of congenital NS. [1] The glomerular phenotype of the disease is associated with other disorders, such as Galloway–Mowat syndrome, Pierson syndrome, Frasier syndrome, and others. The pathology of the kidneys displays prominent, closely spaced podocytes in the early stages, and the glomeruli might be enlarged and exhibit hyaline casts. In immunofluorescence, nonspecific mesangial IgM, C3, and C1q deposits are found, while electron microscopy also shows podocyte hypertrophy and irregular FP effacement. In the late stages of DMS, the glomeruli are damaged and have thickened capillary loops, smaller capillary lumens, and a sclerotic mass in the mesangium [18].
2.3.3. Focal Segmental Glomerular Sclerosis
Focal segmental glomerular sclerosis (FSGS) is responsible for 10 to 20% of NS cases in children [2]. The podocyte is the main site of injury in FSGS. The damage occurs through a variety of mechanisms, including immune, toxic, and viral, mechanical injuries, and genetic dysfunction [19]. Injured podocytes are missed in the urinary space, resulting in podocyte effacement. The remaining podocytes hypertrophy and cover the glomerular capillary surface in response to this lack of podocytes. Intracapillary hypertension can cause damage to podocytes and endothelial cells. It can also cause changes in the mesangial cells that lead to sclerosis [20].
In primary FSGS, a circulating factor may be involved in the pathophysiology of NS. It is probably associated with some cytokines that disturb podocyte function [21]; therefore, it can reoccur after kidney transplantation [22]. In fact, the recurrence of idiopathic FSGS occurs in around one-third of patients after transplant [23]. Some molecules have been associated with the recurrence of FSGS, including cardiotrophin-like cytokine factor 1 [24], apoA1b (an isoform of ApoA1) [25], anti-CD40 antibody [26], and serum urine-type plasminogen activator receptor (suPAR) [27]. The association with suPAR is still controversial [28]. However, up to now, no molecule has been definitively established as the circulating factor in all cases of primary FSGS.
More than 50 genes have been presented as potential factors for monogenic forms of FSGS. The group of genes includes those involved in slit diaphragm structure, actin cytoskeleton, and cell-signaling apparatus [29][30][29,30]. FSGS can also be drug-induced, as IFN-α, -β, or -γ therapy has been linked to the occurrence of collapsing glomerulopathy in a case series of 11 subjects [31]. Anabolic steroid abuse has also been related to the development of FSGS. The association may be due to a combination of a direct nephrotoxic effect and adaptive glomerular changes to the increased lean body mass [32].
2.4. Histological Classifications for Focal Segmental Glomerular Sclerosis
2.4.1. Cellular Focal Segmental Glomerular Sclerosis
The description of this variant resembles focal proliferative glomerulonephritis. On light microscopy, segmental hypercellularity and endocapillary proliferation are reported, with a luminal obliteration of capillaries [2]. Severe FP effacement is also common but with intact basement membranes [2][7][2,7]. Furthermore, there is an increase in mesangial and inflammatory cells such as neutrophils, foam cells, and monocytes. Although this variant shows marked podocyte hyperplasia [2], it is the least common one and is thought to be an early stage in the evolution of sclerotic lesions [7].
2.4.2. Primary Focal Segmental Glomerular Sclerosis with Mesangial Hypercellularity
This variant of FSGS reveals mainly mesangial hypercellularity in the non-sclerotic glomeruli, including the presence of IgM and C3 in these glomeruli on immunofluorescence patterns. Electron microscopy shows the classic segmental sclerosing lesions, severe FP effacement without any electron-dense deposits, and podocyte hyperplasia [2].
2.4.3. Familial Focal Segmental Glomerular Sclerosis
Familial FSGS is highly associated with genetic mutations, especially in genes that encode podocyte proteins [7]. Several defects in the proteins podocin (NPHS2 gene, chromosome 1q25-31) and actinin 4 (ACTN4, chromosome 19q13) cause autosomal dominant FSGS [2][18][2,18]. TRPC6 gene mutations lead to dysregulation of the cell cycle machinery with consequent apoptosis [18], while CD2AP mutations alter the bond between SD and the actin cytoskeleton [33]. Recent studies have shown an association between phospholipase C epsilon gene mutations (PLCE1, chromosome 10q23-33) and early onset NS [34]. The familial FSGS can manifest at any age and accounts for approximately 20% of FSGS cases [35].
2.4.4. Secondary Focal Segmental Glomerular Sclerosis
Other diseases may also trigger FSGS in pediatric patients, including IgA nephropathy [36], hereditary nephritis (Alport’s syndrome) [37], and lupus nephritis [38]. These diseases reveal different histological features, which can be traced using immunofluorescence: mesangial hypercellularity and mesangial IgA in IgA nephropathy, basement membrane abnormalities in Alport syndrome, and concurrent positive staining for IgA, IgM, IgG, C3, and C1q (full-house pattern) in lupus nephritis. Adults and children can develop secondary FSGS [2].
2.4.5. Collapsing Glomerulopathy
Severe and quick loss of renal function is highly associated with collapsing glomerulopathy [18]. This variant of FSGS is characterized by an implosive collapse of the capillary loops with alterations of the basement membrane, especially its wrinkling and contraction, and hypertrophy and hyperplasia of podocytes (Figure 3) [2]. There is no mesangial sclerosis, but the proliferating visceral podocytes and parietal epithelial cells fill Bowman’s space, resembling crescents (pseudocrescents) [18]. FP effacement is notorious even in glomeruli without collapsing lesions. In children, mitochondrial abnormalities have been reported to affect glomerular epithelial cells. COQ2 mutations inherited trigger an autosomal-recessive condition, which is the most common genetic variant of these mitochondrial disorders [39]. Ubiquinone replacement therapy is typical to treat this variant, and early recognition may prevent neurologic complications [18].
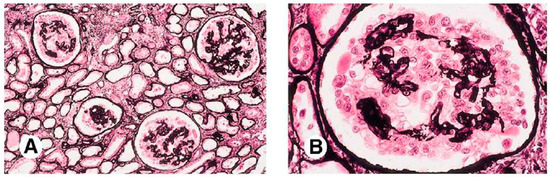
Figure 3. Histological examination of Collapsing Focal Segmental Glomerulosclerosis (cFSGS). (A) Low magnification view (100×) of kidney tissue depicting the characteristic features of collapsing FSGS (cFSGS). The image reveals widespread collapse and obliteration of glomerular capillaries, accompanied by prominent hyperplasia and hypertrophy of podocytes. The collapsing variant is characterized by the involvement of the entire glomerulus, leading to marked changes in the glomerular architecture. (B) Higher magnification (400×) of a representative section illustrating the detailed histopathological changes in cFSGS. At this level of magnification, the severe podocyte hyperplasia, segmental capillary collapse, and visceral epithelial cell hyperplasia become more evident. The presence of prominent protein resorption droplets within dilated tubules is also notable, highlighting the severity of glomerular injury in cFSGS. Both micrographs were obtained using Jones silver staining (personal archive of the authors).
Collapsing glomerulopathy is linked to other causes like infections, drugs, and autoimmune diseases. The original description of this variant occurred in the 1980s during the HIV epidemic, but later it was described in other infections such as malaria and visceral leishmaniasis as well [40]. There is some evidence that other viral infections (cytomegalovirus, parvovirus B19, hepatitis C virus, dengue virus, and Zika virus) and systemic immune responses (tuberculosis-induced collapsing glomerulopathy) target podocytes and their proliferative machinery [41][42][43][44][41,42,43,44]. Patients with this variant are usually steroid-resistant and progress to renal failure quickly [2].
2.4.6. C1q Nephropathy
The description of this form of NS was first written by Jennette and Hipp and is still inconclusive [45]. Histological patterns in C1q nephropathy may vary from MCNS to mild mesangial proliferation and to FSGS. Electron microscopy usually shows mesangial electron-dense deposits, especially immunofluorescence staining for C1q. IgG and/or IgM are commonly present as well [46]. These deposits are predominantly located in paramesangial segments, and FP fusion is also reported. Some light microscopic findings of this entity are similar to the findings seen in lupus nephritis, which is an important differential diagnosis of C1q nephropathy. Furthermore, IgA nephropathy and the membranoproliferative pattern of histology may also distinguish the diagnosis via immunofluorescence. However, additional studies are necessary to understand the pathophysiology of C1q nephropathy and improve diagnosis and treatment [46].
2.5. Hypotheses Linking Glomerulosclerosis to Podocyte-Derived Alterations
The glomerular development is highly regulated with the podocyte, while its altered biology is a determinant of progression to glomerulosclerosis. Different glomerular diseases have been directly associated with alterations in podocyte functions and biology. Therefore, the central role of these specialized and polarized epithelial cells supports the concept of glomerular diseases as podocytopathies [47]. Although podocyte dysfunction, injury, or loss determine a common factor in the development of NS, many hypotheses propose that the pathogenesis of idiopathic NS is either immune-mediated or due to a genetic variant. The presence of a systemic circulating factor, likely podocyte-derived, has also been suggested [1][6][1,6].
2.5.1. Immune-Mediated
The immune system may be involved in the pathogenesis of NS, particularly a dysfunction or dysregulation of T lymphocytes [48]. Some evidence, such as the development of NS after allergic reactions to poisons and stings, reinforces the hypothesis. Furthermore, immunosuppressive agents play a role in NS response and even resolution [1]. In this sense, Lin et al. demonstrated spontaneous NS remission after infection with measles, which triggers prolonged depression of the immune system cells [49]. Chemotherapy for Hodgkin’s and other T-cell lymphomas in NS patients also affects the disease outcome. The literature supports an association between NS, especially MCNS, and classical Hodgkin’s lymphoma (cHL). MCNS associated with cHL is often dependent on or resistant to steroids, but the remission of NS is linked to the cure of the lymphoma [50]. MCNS can also occur in non-Hodgkin lymphoid disorders [51].
The antigen-presenting cells are responsible for the primary co-stimulatory signal for T-cell activation. CD80 (B7-1) is a protein expressed on those antigen-presenting cells and is the main molecular candidate for the immune-mediated feature in podocytopathies. CD80 binds to CTLA-4, a protein receptor expressed on the T-cell surface, and an increase in podocyte B7-1 expression may cause consequent dysfunction or dysregulation of T lymphocytes [52]. Clinical trials tested CTLA-4 as a therapeutic agent, like abatacept [53] and belatacept [54], in FSGS, but results on efficacy are still conflicting.
2.5.2. Systemic Circulating Factors
A hypothetical cause of NS is the existence of a circulating permeability factor that disturbs the glomerular permeability. It has been suggested that this supposed factor acts on the endothelial cell or podocyte, especially in steroid-resistant NS (SRNS) and FSGS [1]. A study developed in 1996 evaluated in vitro the glomerular permeability to albumin of recurrent FSGS patients. The investigation included the response to treatment and recurrence of FSGS after kidney transplantation in 30 to 40 percent of patients. This study sustains the hypothesis that a circulating factor is related to the recurrence of the disease and to the beginning of renal injury [55]. The existence of a circulating factor was further corroborated by the possibility of maternal transmission of FSGS. However, in utero exposure did not cause chronic glomerular disease, suggesting that the supposed factor would be smaller than IgG with a molecular weight between 30 and 50 kd and would not remain present in the fetus’s circulation [56].
Some molecules have been investigated as potential permeability factors, including hemopexin, heparanase, angiopoietin-like 4 (ANGPTL4), cardiotrophin-like cytokine-1, and suPAR [57][58][59][57,58,59]. The suPAR is found in the plasma of patients with recurrent FSGS and may lead to FP effacement and posterior proteinuria due to an interaction with α5β3-integrin receptors on the surface of podocytes [59]. ANGPTL4 is a glycoprotein expressed in the heart, adipose tissue, and skeletal muscle. Its concentration is elevated in NS. ANGPTL4 has two molecular forms, hyposialylated and sialylated, that trigger different mechanisms to regulate proteinuria [58]. The hyposialylated form is secreted via podocytes and affects GBM and endothelial cells, leading to damage to the filtration barrier and proteinuria [58]. On the other hand, the sialylated form binds to α5β3-integrins on the glomerular endothelium and reduces proteinuria. This mechanism has been explored as a potential treatment for NS. However, α5β3-integrin activation is lipid-dependent, and it causes hypertriglyceridemia in NS via inhibition of lipoprotein lipase [58].
2.5.3. Genetic Variants
Mutations on genes encoding some components of the glomerular barrier, such as the GBM, podocyte, its mitochondria or lysosomes, slit diaphragm, actin cytoskeleton, or its molecular composition, can lead to proteinuria and NS. An understanding of the genes related to NS is important for the management of patients, especially considering the genetic variants and mutations associated with SRNS.
The early identification of genetic causes for SRNS allows for more appropriate management of the disease. Specific mutations in the genes NPHS1, NPHS2, LAMB2, and WT1 explain 69 to 85% of NS cases starting in the first three months of life. The presence of mutations in these genes decreases to 50 to 66% when NS starts between 4 and 12 months of life. If NS begins after 1 year of age, only 25% of cases are associated with genetic mutations [60]. The early identification of genetic causes supports the discontinuation of immunosuppressive agents and provides more information for prenatal counseling. In addition, the risk of recurrence after kidney transplant is lower in genetic forms of NS than in non-genetic-related syndromes. A multicentric study followed 1340 children with SRNS over 1 year of age and found that 14% of cases were associated with a genetic mutation [61], including polymorphisms of the major histocompatibility complex (HLA). HLA loci mutations represent a genetic risk for SSNS of 4 to 6 percent. However, the association between SSNS and these mutations is found only in specific ethnicities [62][63][62,63].