Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Alice Bonanni and Version 2 by Peter Tang.
Heart failure with preserved ejection fraction (HFpEF) is a complex and heterogeneous clinical syndrome, and the prevalence is expected to increase in the coming years. This condition poses a burden to the global health care system as the number of patients affected by this condition is constantly increasing due to a rising average lifespan. The absence of validated drugs effective in reducing hospitalization rates and mortality may reflect the impossibility of applying a one size fits all approach as in HFrEF, heading for a personalized approach. Available evidence demonstrated the link between collagen quantity and quality alterations, and cardiac remodeling. In the context of fibrosis, collagen cross-linking is strictly involved, displaying two types of mechanisms: enzymatic and non-enzymatic. In the murine model, enzymatic inhibition of fibrosis-inducing protease-activated receptor-1 (PAR1) and transforming growth factor (TGF)-β signaling appeared to reduce cardiac fibrosis. On the other hand, in the case of non-enzymatic cross-linking, sodium glucose co-transporter type 2 inhibitors (SGLT2is), appeared to counteract the deposition of advanced glycation end-products (AGEs), which in turn contributed to ventricular remodeling.
- HFpEF
- coronary microvascular dysfunction
- cardiac remodeling
- collagen
- SGLT2i
- AGEs
1. Introduction
Heart failure with preserved ejection fraction (HFpEF) is a complex and heterogeneous clinical syndrome that results from heart structural and functional changes, which makes the heart able to adequately pump blood to peripheral districts only at the cost of elevated cardiac filling pressures. This syndrome is characterized by signs and symptoms of HF, such as dyspnea and fatigue, but with a normal left ventricular ejection fraction (LVEF) and, evidence of structural and/or functional cardiac abnormalities consistent with the presence of LV diastolic dysfunction and raised LV filling pressures, according to the latest European Society of Cardiology guidelines [1]. More often diagnosed in women than in men, HFpEF is defined as a “female pattern”, even though it may soon be overcome because of the increasing diagnoses in men [2]. Due to its heterogeneous etiology, HFpEF embraces different phenotypes, sometimes showing a more fibrotic biological profile, while, on other occasions, displaying a more marked inflammatory identity [3]. This condition represents a burden on the health care system worldwide as the number of HFpEF patients is steadily increasing—firstly, due to a rise in the average life expectancy, and secondly, thanks to an easier clinical recognition—remaining one of the most challenging hot topics in the cardiovascular field. Indeed, because of its nature, the scientific community still lacks a perfect experimental animal model that mimics this complex condition, making drug development harder.
2. Endothelial Dysfunction and the HFpEF Microvascular Paradigm
Endothelial dysfunction plays a pivotal role in the pathophysiology of HFpEF [4][7]. Cardiovascular comorbidities, such as hypertension, atrial fibrillation, coronary artery disease, dyslipidemia, diabetes mellitus, obesity and obstructive sleep apnea, trigger a low-grade systemic pro-inflammatory state that leads to coronary microvascular dysfunction (CMD) and affects the perivascular environment, causing fibrosis and remodeling of the heart. The latest updates in biomarker selection highlight the enormous complexity of CMD molecular pathways [5][8]. Among non-cardiovascular comorbidities, obesity elicits a chronic, low-grade systemic inflammatory state in HFpEF patients, which contributes to CMD through adipokine release and β-adrenergic receptor activation. Furthermore, obesity could be an indirect goal to stimulate other comorbidities, such as insulin resistance [6][9]. Diabetes mellitus also fuels the systemic inflammatory milieu, contributing to endothelial impairment. As in patients presenting with myocardial infarction [7][10], the concomitant metabolic dysregulation is characterized by an increase in glucose uptake and related glucose transporter 1 (GLUT-1). In the murine model, this condition is also typical of adverse LV remodeling, cardiomyocyte hypertrophy, and eventually a failing heart [8][11]. Hyperglycemia induces the glycation of interstitial proteins and oxidative stress leads to the formation of advanced glycation end products (AGEs), responsible for the increased LV stiffness. The transition from glucose utilization to the fatty acids-dependent state, lipotoxicity, and impaired insulin signaling, all contribute to HF in the diabetic setting [9][10][12,13].3. Fibrosis and Extracellular Matrix Derangement in the Onset of HFpEF Stiffness
Fibrosis derives from the interplay of modifications that affect the cellular and extracellular microenvironment. The main changes concern cardiomyocyte hypertrophy, alteration of the giant sarcomeric myoprotein titin [11][17]—a bidirectional spring whose phosphorylation state affects cardiomyocyte passive stiffness—and excessive deposition of extracellular matrix (ECM). ECM alteration shapes a scaffold between cardiomyocytes and cardiac vessels leading to an imbalance of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPS) [12][18]. Furthermore, the myocardial ECM not only provides a mere mechanical support that preserves the geometry of the heart but constitutes a dynamic entity, whose turnover relies on fibroblasts which, in a pathological milieu, undergo a conversion towards collagen-producing myofibroblasts. MMPs are in a latent state and their activation can be induced by ROS generation, leading to the degradation of ECM components. MMP1, MMP8 and MMP13 have a high affinity for collagen, while other MMPs have tropism for elastin and proteoglycans [13][16]. Mainly elastin, but also laminin, glycoproteins, and various collagen types, were reported as sources of ECM matrikines. Matrikines, ECM-derived peptides, may regulate ECM synthesis and remodeling and MMP generation and activation [14][19]. Furthermore, the impaired removal of these fragments is involved in the activation of the immune-mediated response, fueling ECM degradation, and further contributing to the disruption of myocardial integrity [15][20]. Selective enzymes, such as MMP-2, MMP-7, TIMP-1 and TIMP-2 increase with age, while MMP-9 shows the opposite trend. Both MMP and TIMP protein levels are augmented and seem to be associated with diastolic impairment in aged and healthy individuals, although more studies are required to further clarify their role in age-related ECM modifications [16][17][21,22]. In this general scenario, cardiomyocyte stiffening and interstitial fibrosis, deriving from collagen deposition and degradation imbalance, lead to LV diastolic dysfunction, the major impairment in HFpEF patients.4. Qualitative and Quantitative Changes of Cardiac Collagen Fibers
Collagen is a vital, life-long and highly prevalent structural protein in mammals. The collagen family embraces 28 members, each consisting of 3 polypeptide subunits that assemble into a triple-helix structure domain [18][23]. Fibrillary collagen type I and III are the prevailing components of the ECM, accounting for nearly 80% and 10% of collagen in the healthy heart, respectively [19][24]. Under physiological conditions, triple helices generate microfibrils which, once assembled together, form larger fibers, increasing tissue stability [18][23]. Previous studies have shown a significant sex-related alteration in collagen metabolism, especially during the menopausal and postmenopausal phases, confirming the female HFpEF pattern and period [20][25]. Indeed, with aging, the collagen properties change, damaging its biochemical and biomechanical characteristics [21][26], thus leading to the loss of flexibility and altered enzymatic digestibility [22][27]. Age-related alterations in collagen strands play a central role in tissue stiffening. Several studies have proven, over the years, the reduction of the collagen fiber number in elderly people [23][24][28,29]. Furthermore, elderly adult collagen net is defined by fewer, longer, and thicker fibrils than the young-adult counterpart [23][28], highlighting the relevance of collagen quantity and quality. Quality features, such as collagen isoforms, collagen cross-linking (CCL) degree, and collagen type I to type III ratio and solubility, also play a role in diastolic impairment [12][18]. In HF related to pressure overload, the ratio of collagen type I to type III is increased [25][30] and is due to the augmented expression of type I collagen, which leads to an imbalance between myocardial stiffness and elasticity, properties respectively provided by the collagen isoform I and isoform III. Nevertheless, in ischemic cardiomyopathy -main cause of HFrEF-, the same index appears reduced, attributable to an increased expression of type III collagen. In this perspective, the collagen type ratio together with different collagen isoforms could be exploited as intriguing tools to distinguish between the two HF phenotypes [26][31] and to tailor therapeutic treatment. For instance, Graziani et al. hypothesized parallelism, transposing therapeutic strategies from idiopathic pulmonary fibrosis to HFpEF, targeting collagen production, induced by transforming growth factor-beta (TGF-β) signaling pathway, adopting nintedanib and pirfenidone [27][32]; thus, updating the possible new applications of these pharmacological treatments (Figure 1).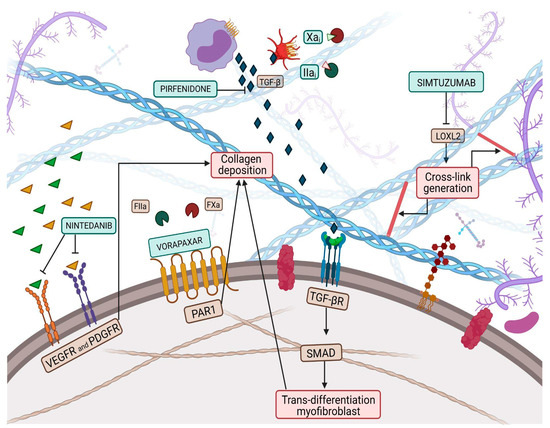
Figure 1. Collagen deposition and collagen cross-link (CCL) have a great influence on left ventricular (LV) stiffness in patients with heart failure (HF). CCL is the formation of intramolecular and intermolecular covalent bonds between lysine residues in collagen molecules, which greatly increases the tensile strength and stiffness of collagen fibers and makes them more resistant to degradation. Multiple pathways have been targeted by therapeutic pharmacological treatments to overcome myofibroblast production, such as nintedanib, pirfenidone, Xa and Iia inhibitors, while vorapaxar attempts to inhibit excessive collagen deposition. Finally, simtuzumab inhibits the lysyl oxidase 2 (LOX2) pathway, thus reducing collagen cross-link generation.