MYC amplification or overexpression is most common in Group 3 medulloblastomas and is positively associated with poor clinical outcomes. Protein arginine methyltransferase 5 (PRMT5) overexpression has been shown to be associated with tumorigenic MYC functions in cancers, particularly in brain cancers such as glioblastoma and medulloblastoma. PRMT5 regulates oncogenes, including MYC, that are often deregulated in medulloblastomas. However, the role of PRMT5-mediated post-translational modification in the stabilization of these oncoproteins remains poorly understood. The potential impact of PRMT5 inhibition on MYC makes it an attractive target in various cancers. PRMT5 inhibitors are a promising class of anti-cancer drugs demonstrating preclinical and preliminary clinical efficacies.
- brain cancer
- medulloblastoma
- MYC
- PRMT5 inhibitors
- SDMA
1. Introduction
2. PRMT5 Structure, Function, and Localization
2.1. Structure
PRMT5 is a primary type II arginine methyl transferase that forms a prominent methylosome complex with distinctive binding oligopeptides, such as the WD (Trp-Asp) repeat-containing 50-kilodalton methylosome protein (MEP50). PRMT5 requires the existence of diverse substrate adapters such as rio-domain-containing protein 1 (RioK1), chloride channel nucleotide-sensitive 1A protein (pIC1n) and cooperator of PRTM5 (COPR5) to detect and catalyze the SDMA on histone and non-histone proteins via PTMs [31,32,33][31][32][33]. PRMT5′s structure consists of a triphosphate isomerase (TIM) barrel, an intermediate Rossmann-fold, and a C-terminal β-barrel [34]. Four PRMT5 units generate a hetero octameric complex by binding with four MEP50s (Figure 1). Studies have demonstrated that PRMT5 alone has minimal methyltransferase activity; it must be complexed with MEP50 to achieve normal catalysis of SDMA on proteins [35]. This could be because MEP50 enhances the stability of PRMT5 for a long time by binding with proteins and acting as a metastable cofactor.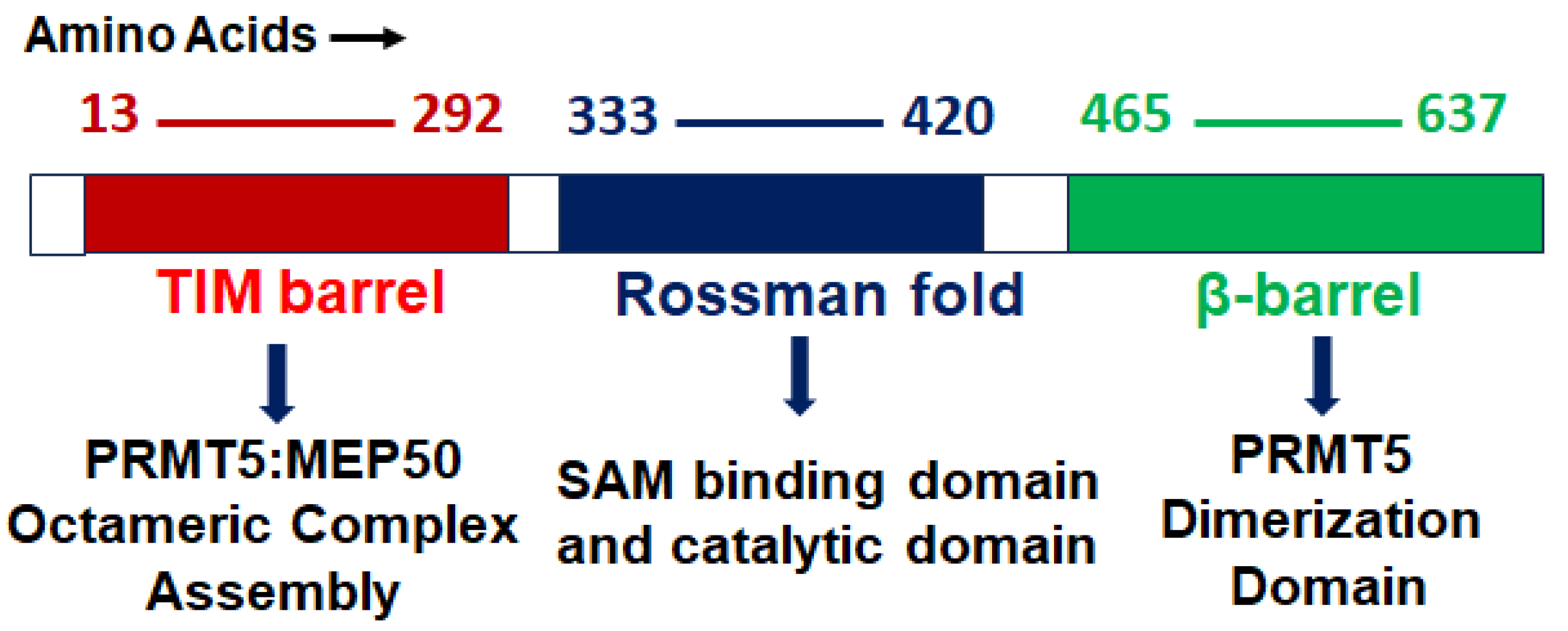
2.2. Function
PRMT5 plays a key role in cell functions and processes by regulating the methylation of cellular proteins, which affects oncogenic cellular processes such as cell proliferation and differentiation [29,30,36][29][30][36]. PRMT5 regulates these processes by modifying gene expression to stabilize histones H4R3, H3R2, H3R8, and H2AR3 and non-histone proteins via the SDMA process [37,38][37][38]. An extensive range of nonhistone proteins have also been revealed as PRMT5 substrates, including androgen receptor (AR), EGFR, GATA4, C-MYC, N-MYC, IL-2, E2F1, GM130, HOXA9, KLF4, KLF5, NOTCH, NFkB(p65), PDCD4, POLR2A, P53, RAF proteins, SPT5, SREBP1a, Sm proteins, nucleolin, and others [12,36,39,40,41,42,43,44,45,46,47,48,49,50,51,52][12][36][39][40][41][42][43][44][45][46][47][48][49][50][51][52]. In addition, some substrates such as certain nonhistone oncogenic transcription factors are symmetrically dimethylated by PRMT5. PRMT5-regulated cellular processes are shown in Figure 2. The importance of arginine methylation by PRMT5 in cancer progression has only recently become apparent [17]. PRMT5 knockout mice exhibited embryonic lethality, demonstrating the role of PRTM5 in embryonic development and crucial biological functions. In mouse embryonic stem cells (ESCs), PRMT5 maintains pluripotency, whereas in human ESCs, it influences only proliferation [53,54][53][54]. PRMT5 is needed for neural stem cell persistence and its deletion causes premature death of the mouse by disrupting the development of the central nervous system [55]. PRMT5 promotes SWI/SNF-mediated chromatin remodeling and controls the process of myogenesis. Deletion of PRMT5 causes an obstacle in developmental processes, uncontrolled proliferation, and impairment of adult tissue differentiation [53,54,55,56,57,58][53][54][55][56][57][58]. Notably, PRMT5 is overexpressed in a number of cancers, including melanoma, multiple myeloma, lymphoma, glioblastoma, breast, lung, pancreas, prostate, ovarian, and colorectal cancers, and high expression of PRMT5 often correlates with poor patient clinical outcomes [38,59][38][59]. Organ-specific functions of PRMT5 are shown in Table 1. The higher expression of PRMT5 in cancer is thought to epigenetically suppress tumor suppressor and cell cycle genes [17,60][17][60]. Recently, the association of PRMT5 with MYC was found in numerous cancers, including brain tumors such as glioblastoma; this association creates abnormalities in MYC function [61,62,63][61][62][63]. Consequently, PRMT5 has been recognized as an oncogenic function and has received extensive interest as a potential target for better clinical outcomes. To this end, numerous potent therapeutic agents have been developed to inhibit PRMT5 and their antitumor effects are now being assessed in preclinical models and clinical trials [27,64][27][64].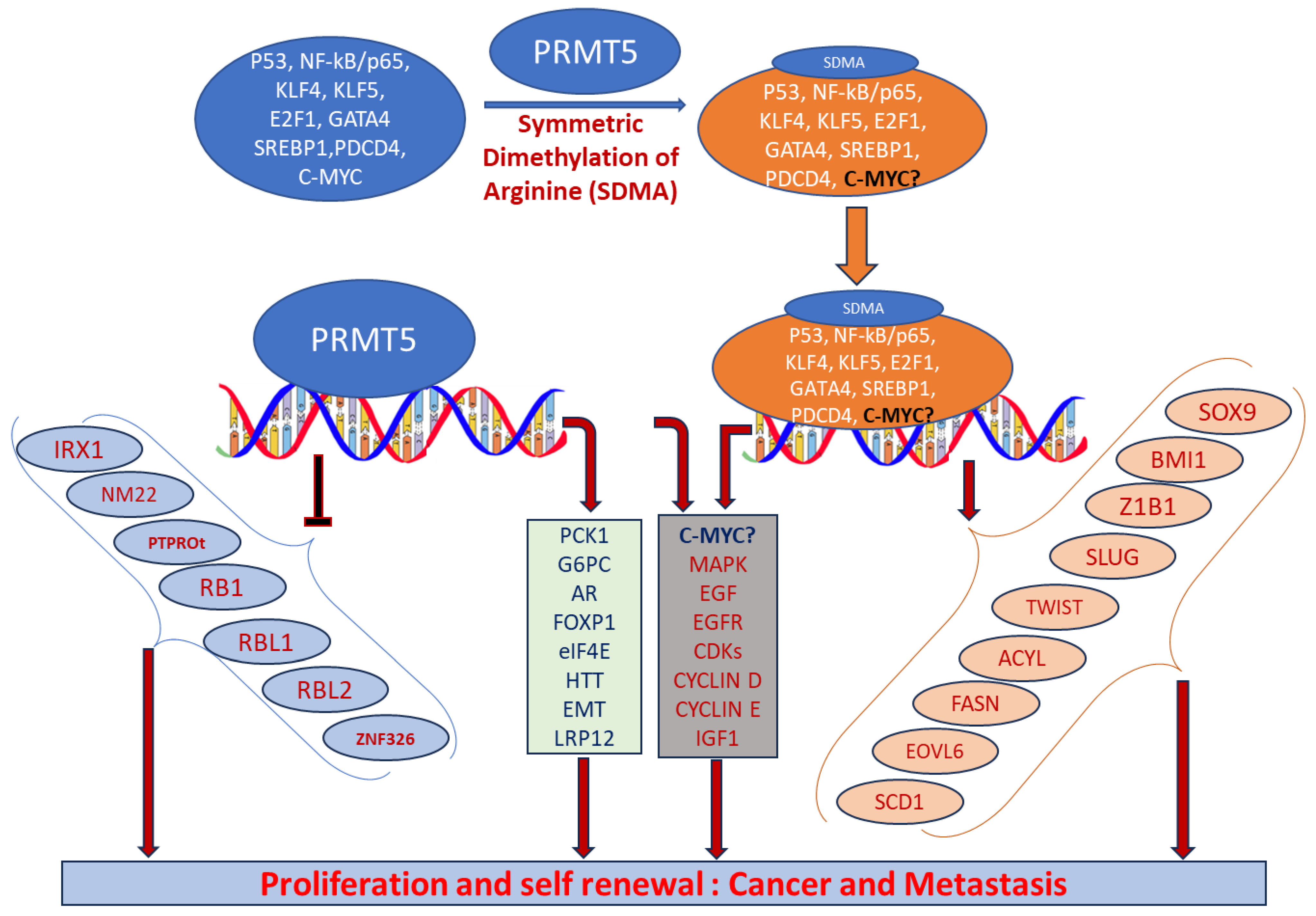
2.3. Localization
Cytosolic and nuclear localization of PRMT5 helps to determine its role in the cell. PRMT5 is predominantly localized in the cytoplasm in lung [65], prostate [66], and melanoma cancer [67]. Diffused cellular localization of PRMT5 was confirmed in both the cytoplasm and the nucleus of brain tumor glioblastoma cells [61]. Cytoplasmic and nuclear localization of PRMT5 has also been confirmed in various preclinical mouse models and primary human cancer tissues [68]. In adult mice, PRMT5 is expressed predominantly in the nucleus of the neurons in the cerebrum and spinal cord [55]. Han et al. demonstrated the high expression of PRMT5 as a marker of malignant progression in glioblastoma and its crucial role in tumor growth [63]. The Ourresearchers' lab recently analyzed the localization of the PRMT5 in tumor tissues of medulloblastoma patients as well as in MYC-amplified cell lines. PRMT5 demonstrated predominantly nuclear localization in both HD-MB03 and primary tumor cells [22].|
Organ |
Cellular Function |
Mechanism |
References |
|---|---|---|---|
|
Brain |
|
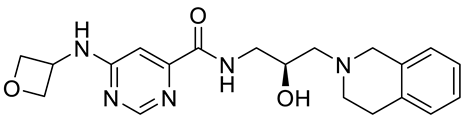
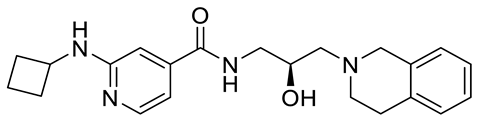
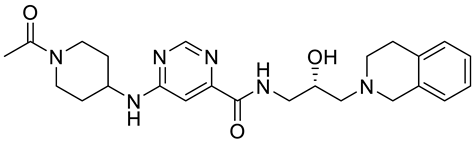
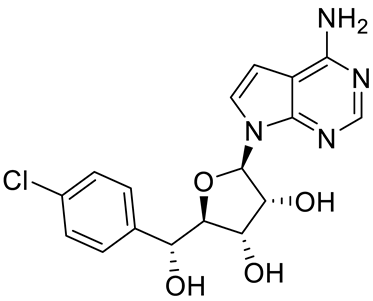
Compound Name |
Structure |
Function |
|---|
|
ClinicalTrials.gov Identifier |
Name of Inhibitor |
IC50 In Vitro |
In Vivo Activity |
References |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
Status |
Disease |
|||||||||
|
Cell cycle progression, apoptosis |
||||||||||
|
JNJ64619178 |
Altered expression and stability of MYC |
[ |
Dual SAM/substrate competitive 22] |
|||||||
0.2 nM | Antitumor effect in lung cancer, AML, non-Hodgkin lymphoma cell line mouse xenograft |
|||||||||
|
NCT03573310 | [ |
JNJ64619178 | ] |
|||||||
Phase I |
Neoplasm solid tumors, non-Hodgkin lymphoma, and myelodysplastic syndrome |
PF06939999 Phase separation |
Methylation of FUS |
SAM competitive [69] |
||||||
3.3 nM | Antitumor effect in lung cancer |
|||||||||
|
NCT03854227 |
PF06939999 | [ | ] |
|||||||
Phase I |
Advance and metastatic solid tumors |
GSK3235025 EPZ015666 GSK3β-NF-kβ signaling |
|
Substrate competitive |
22 nM |
Antitumor effect in MCL, MM, AML, GBM, and bladder cell line mouse xenografts and in a TNBC PDX mouse model |
||||
|
NCT03614728 |
GSK3326595 Altered expression of E2F1 |
[70] |
||||||||
] | [ | ] | [ | |||||||
Phase I and II |
Metastatic solid tumors and acute myeloid leukemia |
GSK591 (EPZ015866) HTT toxicity |
Altered expression of HTT |
[ |
Substrate competitive 71] |
|||||
4 nM | Antitumor effect in glioblastoma |
|||||||||
|
NCT02783300 |
GSK3326595 |
Phase I |
Solid tumors and non-Hodgkin lymphoma | [110] |
||||||
|
GSK3326595 |
AKT-ERK signaling, cell cycle progression |
Altered expression of PTEN |
||||||||
|
NCT04676516 | [72] |
|||||||||
Substrate competitive |
GSK3326595 |
Phase II 6.2 nM |
Early-stage breast cancer Antitumor effect in non-Hodgkin lymphoma cell line mouse xenograft and antitumor effect in a DLBCL PDX mouse model |
Cell cycle progression, stemness |
Altered RNA Splicing |
|||||
|
AMG 193 |
||||||||||
|
NCT03886831 |
Structure undisclosed |
PRT543 MTA cooperative inhibitor |
NA |
Phase I ] |
||||||
Antitumor effect on advanced/metastatic solid tumors | [119] |
|||||||||
Advanced solid tumors and hematological malignancies |
PRT543 mTOR signaling |
Methylation (hnRNPA1) |
[75] |
|||||||
SAM |
competitive |
10.8 nM |
Antitumor effect on advanced solid tumors and hematologic malignancies |
|||||||
|
NCT05275478 | [ | ] | ||||||||
TNG908 |
Phase I and II (recruiting) |
Locally advanced solid tumors |
DNA instability response |
Altered expression of RNF168 |
[ |
PRT38276] |
||||
|
NCT04089449 |
Structure undisclosed |
PRT811 SAM competitive |
Phase I (recruiting) 2.8 nM |
Antitumor effect on hematological tumors |
[85] |
|||||
Advanced solid tumors, recurrent glioma, and CNS lymphoma |
PRT811 Cell migration, cell cycle progression, and apoptosis |
Altered expression of LRP12 |
||||||||
|
NCT05245500 | Structure undisclosed |
MRTX1719 SAM competitive | [62] |
|||||||
Phase I and II (recruiting) |
3.9 nM |
Antitumor effect on advanced solid tumor, Glioblastoma, CNS Lymphoma |
[121] |
AKT signaling and metastasis | ||||||
Mesothelioma, NSCLC, malignant peripheral nerve sheath tumors, solid tumors, and pancreatic adenocarcinoma |
Methylation of PKB |
[77] |
||||||||
|
TNG908 |
||||||||||
|
NCT05094336 |
Structure undisclosed |
AMG 193 MTA cooperative inhibitor |
110 nM |
Phase I and II (recruiting) |
Antitumor effect on Glioblastoma, |
[122] |
||||
Advanced MTAP-null solid tumors |
Lungs |
MRTX1719 Metastasis |
Altered expression of EMT genes |
PRMT5–MTA complex inhibitor, MTA competitive [78] |
||||||
12 nM | Antitumor effect on solid tumor |
[ | ||||||||
|
NCT05528055 |
SCR6920 |
Phase I (recruiting) | , | |||||||
Advanced malignant tumors |
LLY-283 (C220) Metastasis |
Altered expression of SHARPIN |
[79 |
|||||||
|
NCT05015309 |
| ] | ||||||||
SH3765 |
SAM |
Phase I (not yet Recruiting) competitive |
22 nM |
Reduced acute graft versus host disease incidence in mice, antitumor effect in MPN xenografts |
Advanced malignant tumors |
|||||
|
Compound1a |
Metastasis |
Altered expression of FGFR3/miR-99 family |
Allosteric modulator [80] |
|||||||
16 nM | Antitumor effect in breast cancer |
[127] |
Metastasis |
Methylation of KLF5 |
[81] |
|||||
|
CMP5 |
Structure undisclosed |
SAM competitive |
25 µM |
Antitumor effect in breast cancer and glioblastoma |
[72] |
Liver |
Lipid metabolism |
|||
|
JBI-778 |
Structure undisclosed | Methylation of SREBP |
Substrate competitive [47] |
|||||||
27 to 700 nM |
Antitumor effect in glioblastoma |
[128] |
ERK signaling |
Altered expression of BTG2 |
[82] |
|||||
|
PRMY5 deprivation |
||||||||||
|
SH3765 |
Structure undisclosed |
Substrate competitive |
NA |
Antitumor effect on advanced malignant tumors, including solid tumors and non-Hodgkin lymphoma |
[PRMT5 activity of LINC01138 |
[83] |
||||
] | ||||||||||
|
SCR6920 |
Structure undisclosed |
WNT-β-Catenin signaling |
Altered cofactor binding of LYRIC |
[84] |
||||||
Substrate competitive | NA |
Antitumor effect on advanced malignant tumor including solid tumor and non-Hodgkin lymphoma |
[129] |
Spleen |
NA |
Altered stability of MYC |
[85] |
|||
|
Pancreas |
Glucose metabolism, cell cycle progression |
Altered stability of MYC |
||||||||
|
Bone |
Type I interferon signaling |
Altered expression (interferon gene) |
||||||||
|
Prostate |
AR, ERG signaling |
Altered methylation (AR) |
||||||||
|
Ovary |
NA |
Altered methylation (E2F1) |
[92] |
|||||||
|
Heart |
Transcriptional activity |
Methylation (GATA4) |
[45] |
|||||||
|
Breast |
Stemness |
Altered expression of C-MYC, KLF4, and OCT4 |
[93] |
|||||||
|
Stemness |
Altered expression of FOXP1 |
[94] |
||||||||
|
Metastasis and invasion |
Altered expression of AKT genes |
[78] |
||||||||
|
Metastasis and AKT signaling |
Methylation of AKT |
[95] |
||||||||
|
Cell cycle progression |
Methylation of KLF4 |
[96] |
||||||||
|
Cell migration |
Methylation of ZNF326 |
[97] |
||||||||
|
NA |
Methylation of PDCD4 |
[98] |
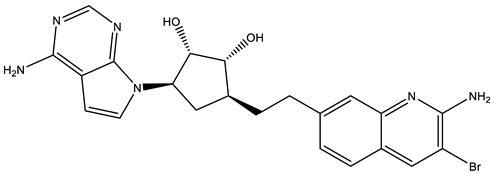
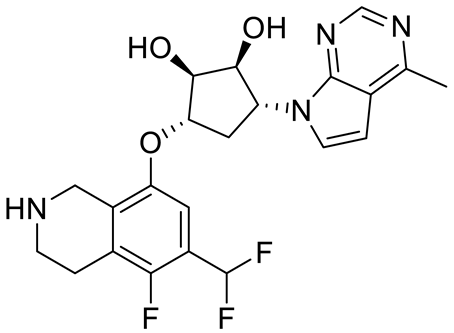
Abbreviations: AKT-ERK, alpha serine/threonine-protein-extracellular-regulated kinase; AR, androgen receptor; E2F1, E2 promoter binding factor 1; EMT, epithelial–mesenchymal transition; ERG, ETS-related gene; FGFR3, fibroblast growth factor 3; FOXP1, forkhead box protein P1; FUS, fused in sarcoma; GSK3β-NF-kβ, glycogen synthase kinase; hnRNPA1, heterogeneous nuclear ribonucleoprotein A1; HTT, huntingtin protein; KLF4, Kruppel-like factor 4; LRP12, low-density lipoprotein receptor-related protein 12; LINC01138, long non-coding RNA; miR-99, microRNA99; mTOR, mammalian target of rapamycin; OCT4, octamer binding protein 4; PKB, protein kinase B; PDCD4, program cell death protein 4; SREBP, sterol regulatory element-binding protein; ZNF326, zinc finger protein 326.
3. PRMT5 Association with MYC-Driven Medulloblastoma
Epigenetic deregulation plays a key role in medulloblastoma tumorigenesis, especially in aggressive Group 3 and Group 4 medulloblastomas [99[99][100][101][102],100,101,102], where germline mutations in known cancer predisposition genes are rare. Indeed, epigenetic deregulator or chromatin modifiers, including histone acetylase or methylation/methyltransferase activities, are very common in Group 3 and 4 medulloblastomas compared to other subgroups. This emphasizes the need to discover and understand the pertinent mechanisms of epigenetic regulation or PTMs and the corresponding therapeutic targets. WThe researchers recently reported that PRTM5 is a critical regulator MYC oncoprotein in an MYC-amplified (Group 3) medulloblastoma [22]. WThe researchers found that high levels of PRMT5 not only mirror MYC expression but also correlate with poor outcomes in Group 3 medulloblastoma patients. Mechanistically, we shthe researchers showed that PRMT5 stabilizes the MYC protein by physically interacting with it, raising the intriguing possibility that PRMT5 can regulate MYC function at both the transcriptional and translational/post-translational levels. The exact MYC oncogenic programs regulated by PRMT5 in medulloblastoma are largely unknown. Therefore, exploring the regulation of MYC-driven oncogenic progresses by PRMT5 is crucial to identify effective therapeutics for these high-risk patients. The involvement of PRMT5 has been verified in the epigenetic regulation of chromatin complexes following interaction with numerous proteins, including transcription factors [42], and their activities are dysregulated in various cancers [59]. In recent studies, high levels of PRMT5 and MYC corelate with glioma malignancy [61,62,63][61][62][63]. Furthermore, PRMT5 is physically associated with N-MYC (an MYC homologue) and enhances the stability of N-MYC in neuroblastoma cells [51]. Nonetheless, the function of PRMT5 and its interaction with MYC in MYC-driven medulloblastoma have not been fully investigated. Favia et al. reported that the association of PRMT1 and PRMT5 with MYC in glioblastoma stem cells resulted in MYC being dimethylated symmetrically and asymmetrically by both enzymes, respectively [103]. MYC-driven cellular processes resulting from symmetric dimethylation by PRMT5 are shown in Figure 3. The colocalization of PRMT5 and MYC suggests that PRMT5 forms a complex with MYC and supports its stabilization in MYC-amplified medulloblastoma cells. This physical interaction of PRMT5 and MYC implies a potential role of PRMT5 in medulloblastoma tumorigenesis.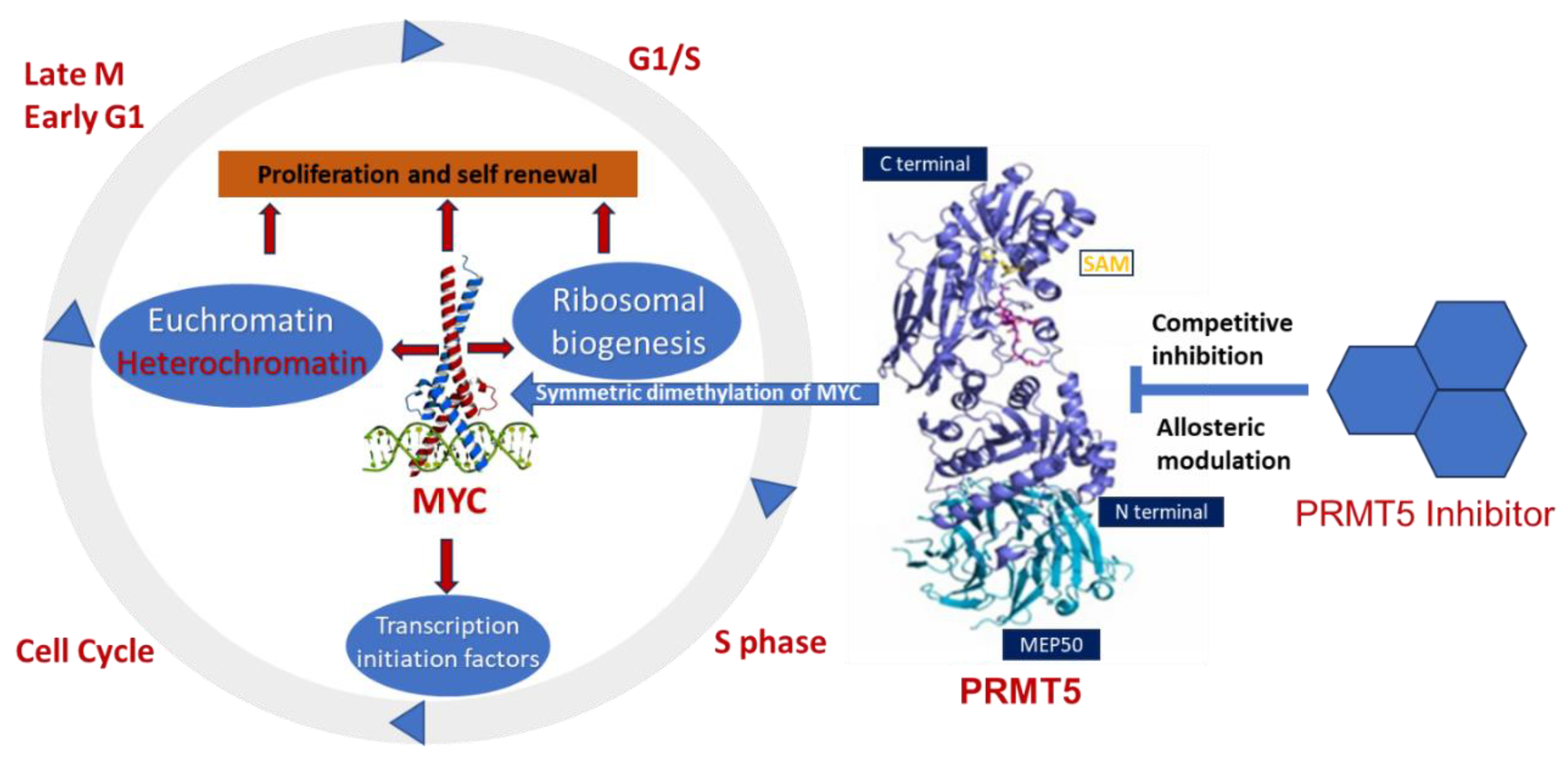
4. Potential Inhibitors of PRMT5
Abbreviations: AML, acute myeloid leukemia; MCL, mantle cell lymphoma; MM, myelomonocytic leukemia; GBM, glioblastoma; TNBC, triple-negative breast cancer, PDX, patient-derived xenograft; DLBCL, diffuse large B cell lymphoma; CNS, central nervous system; MPN, myeloproliferative neoplasm; nM, nano molar; NA, not available; SAM, S-adenosylmethionine; MTA, methylthioadenosine.
References
- Packer, R.J.; Macdonald, T.; Vezina, G.; Keating, R.; Santi, M. Medulloblastoma and primitive neuroectodermal tumors. Handb. Clin. Neurol. 2012, 105, 529–548.
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754.e736.
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Grobner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317.
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472.
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.J.; Koster, J.; Schouten-van Meeteren, A.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484.
- Min, H.S.; Lee, J.Y.; Kim, S.K.; Park, S.H. Genetic grouping of medulloblastomas by representative markers in pathologic diagnosis. Transl. Oncol. 2013, 6, 265–272.
- Juraschka, K.; Taylor, M.D. Medulloblastoma in the age of molecular subgroups: A review. J. Neurosurg. Pediatr. 2019, 24, 353–363.
- Gajjar, A.J.; Robinson, G.W. Medulloblastoma-translating discoveries from the bench to the bedside. Nat. Rev. Clin. Oncol. 2014, 11, 714–722.
- Cho, Y.J.; Tsherniak, A.; Tamayo, P.; Santagata, S.; Ligon, A.; Greulich, H.; Berhoukim, R.; Amani, V.; Goumnerova, L.; Eberhart, C.G.; et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J. Clin. Oncol. 2011, 29, 1424–1430.
- Hill, R.M.; Kuijper, S.; Lindsey, J.C.; Petrie, K.; Schwalbe, E.C.; Barker, K.; Boult, J.K.; Williamson, D.; Ahmad, Z.; Hallsworth, A.; et al. Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell 2015, 27, 72–84.
- Pei, Y.; Moore, C.E.; Wang, J.; Tewari, A.K.; Eroshkin, A.; Cho, Y.J.; Witt, H.; Korshunov, A.; Read, T.A.; Sun, J.L.; et al. An animal model of MYC-driven medulloblastoma. Cancer Cell 2012, 21, 155–167.
- MacDonald, T.J.; Rood, B.R.; Santi, M.R.; Vezina, G.; Bingaman, K.; Cogen, P.H.; Packer, R.J. Advances in the diagnosis, molecular genetics, and treatment of pediatric embryonal CNS tumors. Oncologist 2003, 8, 174–186.
- Zhou, M.; Jiang, J. Gli Phosphorylation Code in Hedgehog Signal Transduction. Front. Cell Dev. Biol. 2022, 10, 846927.
- Zhang, Q.; Jiang, J. Regulation of Hedgehog Signal Transduction by Ubiquitination and Deubiquitination. Int. J. Mol. Sci. 2021, 22, 13338.
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142.
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24.
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50.
- Smith, E.; Zhou, W.; Shindiapina, P.; Sif, S.; Li, C.; Baiocchi, R.A. Recent advances in targeting protein arginine methyltransferase enzymes in cancer therapy. Expert. Opin. Ther. Targets 2018, 22, 527–545.
- Wolf, S.S. The protein arginine methyltransferase family: An update about function, new perspectives and the physiological role in humans. Cell Mol. Life Sci. 2009, 66, 2109–2121.
- Musiani, D.; Bok, J.; Massignani, E.; Wu, L.; Tabaglio, T.; Ippolito, M.R.; Cuomo, A.; Ozbek, U.; Zorgati, H.; Ghoshdastider, U.; et al. Proteomics profiling of arginine methylation defines PRMT5 substrate specificity. Sci. Signal. 2019, 12, eaat8388.
- Abe, Y.; Suzuki, Y.; Kawamura, K.; Tanaka, N. MEP50/PRMT5-mediated methylation activates GLI1 in Hedgehog signalling through inhibition of ubiquitination by the ITCH/NUMB complex. Commun. Biol. 2019, 2, 23.
- Chaturvedi, N.K.; Mahapatra, S.; Kesherwani, V.; Kling, M.J.; Shukla, M.; Ray, S.; Kanchan, R.; Perumal, N.; McGuire, T.R.; Sharp, J.G.; et al. Role of protein arginine methyltransferase 5 in group 3 (MYC-driven) Medulloblastoma. BMC Cancer 2019, 19, 1056.
- Wynn, D.T.; Rodriguez-Blanco, J.; Long, J.; Yang, F.; Shen, C.; Fei, D.; Tang, H.Y.; Hanson, D.; Robbins, D.J. Protein arginine methyltransferase 5 regulates SHH-subgroup medulloblastoma progression. Neurooncol. Adv. 2022, 4, vdac144.
- Pal, S.; Baiocchi, R.A.; Byrd, J.C.; Grever, M.R.; Jacob, S.T.; Sif, S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 2007, 26, 3558–3569.
- Wang, L.; Pal, S.; Sif, S. Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol. Cell Biol. 2008, 28, 6262–6277.
- Koh, C.M.; Bezzi, M.; Low, D.H.; Ang, W.X.; Teo, S.X.; Gay, F.P.; Al-Haddawi, M.; Tan, S.Y.; Osato, M.; Sabo, A.; et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 2015, 523, 96–100.
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437.
- Brehmer, D.; Beke, L.; Wu, T.; Millar, H.J.; Moy, C.; Sun, W.; Mannens, G.; Pande, V.; Boeckx, A.; van Heerde, E.; et al. Discovery and Pharmacological Characterization of JNJ-64619178, a Novel Small-Molecule Inhibitor of PRMT5 with Potent Antitumor Activity. Mol. Cancer Ther. 2021, 20, 2317–2328.
- Litzler, L.C.; Zahn, A.; Meli, A.P.; Hebert, S.; Patenaude, A.M.; Methot, S.P.; Sprumont, A.; Bois, T.; Kitamura, D.; Costantino, S.; et al. PRMT5 is essential for B cell development and germinal center dynamics. Nat. Commun. 2019, 10, 22.
- Barczak, W.; Jin, L.; Carr, S.M.; Munro, S.; Ward, S.; Kanapin, A.; Samsonova, A.; La Thangue, N.B. PRMT5 promotes cancer cell migration and invasion through the E2F pathway. Cell Death Dis. 2020, 11, 572.
- Guderian, G.; Peter, C.; Wiesner, J.; Sickmann, A.; Schulze-Osthoff, K.; Fischer, U.; Grimmler, M. RioK1, a new interactor of protein arginine methyltransferase 5 (PRMT5), competes with pICln for binding and modulates PRMT5 complex composition and substrate specificity. J. Biol. Chem. 2011, 286, 1976–1986.
- Krzyzanowski, A.; Gasper, R.; Adihou, H.; Hart, P.; Waldmann, H. Biochemical Investigation of the Interaction of pICln, RioK1 and COPR5 with the PRMT5-MEP50 Complex. Chembiochem 2021, 22, 1908–1914.
- Mulvaney, K.M.; Blomquist, C.; Acharya, N.; Li, R.; Ranaghan, M.J.; O’Keefe, M.; Rodriguez, D.J.; Young, M.J.; Kesar, D.; Pal, D.; et al. Molecular basis for substrate recruitment to the PRMT5 methylosome. Mol. Cell 2021, 81, 3481–3495.e7.
- Fu, S.; Zheng, Q.; Zhang, D.; Lin, C.; Ouyang, L.; Zhang, J.; Chen, L. Medicinal chemistry strategies targeting PRMT5 for cancer therapy. Eur. J. Med. Chem. 2022, 244, 114842.
- Burgos, E.S.; Wilczek, C.; Onikubo, T.; Bonanno, J.B.; Jansong, J.; Reimer, U.; Shechter, D. Histone H2A and H4 N-terminal tails are positioned by the MEP50 WD repeat protein for efficient methylation by the PRMT5 arginine methyltransferase. J. Biol. Chem. 2015, 290, 9674–9689.
- Scoumanne, A.; Zhang, J.; Chen, X. PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res. 2009, 37, 4965–4976.
- Wei, H.; Mundade, R.; Lange, K.C.; Lu, T. Protein arginine methylation of non-histone proteins and its role in diseases. Cell Cycle 2014, 13, 32–41.
- Stopa, N.; Krebs, J.E.; Shechter, D. The PRMT5 arginine methyltransferase: Many roles in development, cancer and beyond. Cell Mol. Life Sci. 2015, 72, 2041–2059.
- Richard, S.; Morel, M.; Cleroux, P. Arginine methylation regulates IL-2 gene expression: A role for protein arginine methyltransferase 5 (PRMT5). Biochem. J. 2005, 388, 379–386.
- Zhou, Z.; Sun, X.; Zou, Z.; Sun, L.; Zhang, T.; Guo, S.; Wen, Y.; Liu, L.; Wang, Y.; Qin, J.; et al. PRMT5 regulates Golgi apparatus structure through methylation of the golgin GM130. Cell Res. 2010, 20, 1023–1033.
- Hsu, J.M.; Chen, C.T.; Chou, C.K.; Kuo, H.P.; Li, L.Y.; Lin, C.Y.; Lee, H.J.; Wang, Y.N.; Liu, M.; Liao, H.W.; et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat. Cell Biol. 2011, 13, 174–181.
- Karkhanis, V.; Hu, Y.J.; Baiocchi, R.A.; Imbalzano, A.N.; Sif, S. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem. Sci. 2011, 36, 633–641.
- Bandyopadhyay, S.; Harris, D.P.; Adams, G.N.; Lause, G.E.; McHugh, A.; Tillmaand, E.G.; Money, A.; Willard, B.; Fox, P.L.; Dicorleto, P.E. HOXA9 methylation by PRMT5 is essential for endothelial cell expression of leukocyte adhesion molecules. Mol. Cell Biol. 2012, 32, 1202–1213.
- Cho, E.C.; Zheng, S.; Munro, S.; Liu, G.; Carr, S.M.; Moehlenbrink, J.; Lu, Y.C.; Stimson, L.; Khan, O.; Konietzny, R.; et al. Arginine methylation controls growth regulation by E2F-1. EMBO J. 2012, 31, 1785–1797.
- Chen, M.; Yi, B.; Sun, J. Inhibition of cardiomyocyte hypertrophy by protein arginine methyltransferase 5. J. Biol. Chem. 2014, 289, 24325–24335.
- Fay, M.M.; Clegg, J.M.; Uchida, K.A.; Powers, M.A.; Ullman, K.S. Enhanced arginine methylation of programmed cell death 4 protein during nutrient deprivation promotes tumor cell viability. J. Biol. Chem. 2014, 289, 17541–17552.
- Liu, L.; Zhao, X.; Zhao, L.; Li, J.; Yang, H.; Zhu, Z.; Liu, J.; Huang, G. Arginine Methylation of SREBP1a via PRMT5 Promotes De Novo Lipogenesis and Tumor Growth. Cancer Res. 2016, 76, 1260–1272.
- Zhao, D.Y.; Gish, G.; Braunschweig, U.; Li, Y.; Ni, Z.; Schmitges, F.W.; Zhong, G.; Liu, K.; Li, W.; Moffat, J.; et al. SMN and symmetric arginine dimethylation of RNA polymerase II C-terminal domain control termination. Nature 2016, 529, 48–53.
- Fong, J.Y.; Pignata, L.; Goy, P.A.; Kawabata, K.C.; Lee, S.C.; Koh, C.M.; Musiani, D.; Massignani, E.; Kotini, A.G.; Penson, A.; et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 2019, 36, 194–209.e199.
- Checa-Rodriguez, C.; Cepeda-Garcia, C.; Ramon, J.; Lopez-Saavedra, A.; Balestra, F.R.; Dominguez-Sanchez, M.S.; Gomez-Cabello, D.; Huertas, P. Methylation of the central transcriptional regulator KLF4 by PRMT5 is required for DNA end resection and recombination. DNA Repair 2020, 94, 102902.
- Park, J.H.; Szemes, M.; Vieira, G.C.; Melegh, Z.; Malik, S.; Heesom, K.J.; Von Wallwitz-Freitas, L.; Greenhough, A.; Brown, K.W.; Zheng, Y.G.; et al. Protein arginine methyltransferase 5 is a key regulator of the MYCN oncoprotein in neuroblastoma cells. Mol. Oncol. 2015, 9, 617–627.
- Wei, H.; Wang, B.; Miyagi, M.; She, Y.; Gopalan, B.; Huang, D.B.; Ghosh, G.; Stark, G.R.; Lu, T. PRMT5 dimethylates R30 of the p65 subunit to activate NF-kappaB. Proc. Natl. Acad. Sci. USA 2013, 110, 13516–13521.
- Tee, W.W.; Pardo, M.; Theunissen, T.W.; Yu, L.; Choudhary, J.S.; Hajkova, P.; Surani, M.A. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 2010, 24, 2772–2777.
- Gkountela, S.; Li, Z.; Chin, C.J.; Lee, S.A.; Clark, A.T. PRMT5 is required for human embryonic stem cell proliferation but not pluripotency. Stem Cell Rev. Rep. 2014, 10, 230–239.
- Bezzi, M.; Teo, S.X.; Muller, J.; Mok, W.C.; Sahu, S.K.; Vardy, L.A.; Bonday, Z.Q.; Guccione, E. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 2013, 27, 1903–1916.
- Dacwag, C.S.; Bedford, M.T.; Sif, S.; Imbalzano, A.N. Distinct protein arginine methyltransferases promote ATP-dependent chromatin remodeling function at different stages of skeletal muscle differentiation. Mol. Cell Biol. 2009, 29, 1909–1921.
- Li, Z.; Yu, J.; Hosohama, L.; Nee, K.; Gkountela, S.; Chaudhari, S.; Cass, A.A.; Xiao, X.; Clark, A.T. The Sm protein methyltransferase PRMT5 is not required for primordial germ cell specification in mice. EMBO J. 2015, 34, 748–758.
- Zhang, T.; Gunther, S.; Looso, M.; Kunne, C.; Kruger, M.; Kim, J.; Zhou, Y.; Braun, T. Prmt5 is a regulator of muscle stem cell expansion in adult mice. Nat. Commun. 2015, 6, 7140.
- Shailesh, H.; Zakaria, Z.Z.; Baiocchi, R.; Sif, S. Protein arginine methyltransferase 5 (PRMT5) dysregulation in cancer. Oncotarget 2018, 9, 36705–36718.
- Pal, S.; Vishwanath, S.N.; Erdjument-Bromage, H.; Tempst, P.; Sif, S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell Biol. 2004, 24, 9630–9645.
- Mongiardi, M.P.; Savino, M.; Bartoli, L.; Beji, S.; Nanni, S.; Scagnoli, F.; Falchetti, M.L.; Favia, A.; Farsetti, A.; Levi, A.; et al. Myc and Omomyc functionally associate with the Protein Arginine Methyltransferase 5 (PRMT5) in glioblastoma cells. Sci. Rep. 2015, 5, 15494.
- Yan, F.; Alinari, L.; Lustberg, M.E.; Martin, L.K.; Cordero-Nieves, H.M.; Banasavadi-Siddegowda, Y.; Virk, S.; Barnholtz-Sloan, J.; Bell, E.H.; Wojton, J.; et al. Genetic validation of the protein arginine methyltransferase PRMT5 as a candidate therapeutic target in glioblastoma. Cancer Res. 2014, 74, 1752–1765.
- Han, X.; Li, R.; Zhang, W.; Yang, X.; Wheeler, C.G.; Friedman, G.K.; Province, P.; Ding, Q.; You, Z.; Fathallah-Shaykh, H.M.; et al. Expression of PRMT5 correlates with malignant grade in gliomas and plays a pivotal role in tumor growth in vitro. J. Neurooncol. 2014, 118, 61–72.
- Wang, Y.; Hu, W.; Yuan, Y. Protein Arginine Methyltransferase 5 (PRMT5) as an Anticancer Target and Its Inhibitor Discovery. J. Med. Chem. 2018, 61, 9429–9441.
- Ibrahim, R.; Matsubara, D.; Osman, W.; Morikawa, T.; Goto, A.; Morita, S.; Ishikawa, S.; Aburatani, H.; Takai, D.; Nakajima, J.; et al. Expression of PRMT5 in lung adenocarcinoma and its significance in epithelial-mesenchymal transition. Hum. Pathol. 2014, 45, 1397–1405.
- Gu, Z.; Li, Y.; Lee, P.; Liu, T.; Wan, C.; Wang, Z. Protein arginine methyltransferase 5 functions in opposite ways in the cytoplasm and nucleus of prostate cancer cells. PLoS ONE 2012, 7, e44033.
- Nicholas, C.; Yang, J.; Peters, S.B.; Bill, M.A.; Baiocchi, R.A.; Yan, F.; Sif, S.; Tae, S.; Gaudio, E.; Wu, X.; et al. PRMT5 is upregulated in malignant and metastatic melanoma and regulates expression of MITF and p27(Kip1). PLoS ONE 2013, 8, e74710.
- Koh, C.M.; Bezzi, M.; Guccion, E. The Where and the How of PRMT5. Curr. Mol. Bio Rep. 2015, 1, 19–28.
- Chitiprolu, M.; Jagow, C.; Tremblay, V.; Bondy-Chorney, E.; Paris, G.; Savard, A.; Palidwor, G.; Barry, F.A.; Zinman, L.; Keith, J.; et al. A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat. Commun. 2018, 9, 2794.
- Quan, X.; Yue, W.; Luo, Y.; Cao, J.; Wang, H.; Wang, Y.; Lu, Z. The protein arginine methyltransferase PRMT5 regulates Abeta-induced toxicity in human cells and Caenorhabditis elegans models of Alzheimer’s disease. J. Neurochem. 2015, 134, 969–977.
- Ratovitski, T.; Arbez, N.; Stewart, J.C.; Chighladze, E.; Ross, C.A. PRMT5- mediated symmetric arginine dimethylation is attenuated by mutant huntingtin and is impaired in Huntington’s disease (HD). Cell Cycle 2015, 14, 1716–1729.
- Banasavadi-Siddegowda, Y.K.; Russell, L.; Frair, E.; Karkhanis, V.A.; Relation, T.; Yoo, J.Y.; Zhang, J.; Sif, S.; Imitola, J.; Baiocchi, R.; et al. PRMT5-PTEN molecular pathway regulates senescence and self-renewal of primary glioblastoma neurosphere cells. Oncogene 2017, 36, 263–274.
- Braun, C.J.; Stanciu, M.; Boutz, P.L.; Patterson, J.C.; Calligaris, D.; Higuchi, F.; Neupane, R.; Fenoglio, S.; Cahill, D.P.; Wakimoto, H.; et al. Coordinated Splicing of Regulatory Detained Introns within Oncogenic Transcripts Creates an Exploitable Vulnerability in Malignant Glioma. Cancer Cell 2017, 32, 411–426.e411.
- Sachamitr, P.; Ho, J.C.; Ciamponi, F.E.; Ba-Alawi, W.; Coutinho, F.J.; Guilhamon, P.; Kushida, M.M.; Cavalli, F.M.G.; Lee, L.; Rastegar, N.; et al. PRMT5 inhibition disrupts splicing and stemness in glioblastoma. Nat. Commun. 2021, 12, 979.
- Holmes, B.; Benavides-Serrato, A.; Saunders, J.T.; Landon, K.A.; Schreck, A.J.; Nishimura, R.N.; Gera, J. The protein arginine methyltransferase PRMT5 confers therapeutic resistance to mTOR inhibition in glioblastoma. J. Neurooncol. 2019, 145, 11–22.
- Du, C.; Hansen, L.J.; Singh, S.X.; Wang, F.; Sun, R.; Moure, C.J.; Roso, K.; Greer, P.K.; Yan, H.; He, Y. A PRMT5-RNF168-SMURF2 Axis Controls H2AX Proteostasis. Cell Rep. 2019, 28, 3199–3211.e3195.
- Huang, L.; Zhang, X.O.; Rozen, E.J.; Sun, X.; Sallis, B.; Verdejo-Torres, O.; Wigglesworth, K.; Moon, D.; Huang, T.; Cavaretta, J.P.; et al. PRMT5 activates AKT via methylation to promote tumor metastasis. Nat. Commun. 2022, 13, 3955.
- Chen, H.; Lorton, B.; Gupta, V.; Shechter, D. A TGFbeta-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene 2017, 36, 373–386.
- Fu, T.; Lv, X.; Kong, Q.; Yuan, C. A novel SHARPIN-PRMT5-H3R2me1 axis is essential for lung cancer cell invasion. Oncotarget 2017, 8, 54809–54820.
- Jing, P.; Zhao, N.; Ye, M.; Zhang, Y.; Zhang, Z.; Sun, J.; Wang, Z.; Zhang, J.; Gu, Z. Protein arginine methyltransferase 5 promotes lung cancer metastasis via the epigenetic regulation of miR-99 family/FGFR3 signaling. Cancer Lett. 2018, 427, 38–48.
- Zhou, H.; Chang, J.; Zhang, J.; Zheng, H.; Miao, X.; Mo, H.; Sun, J.; Jia, Q.; Qi, G. PRMT5 activates KLF5 by methylation to facilitate lung cancer. J. Cell Mol. Med. 2023, 1–15.
- Jiang, H.; Zhu, Y.; Zhou, Z.; Xu, J.; Jin, S.; Xu, K.; Zhang, H.; Sun, Q.; Wang, J.; Xu, J. PRMT5 promotes cell proliferation by inhibiting BTG2 expression via the ERK signaling pathway in hepatocellular carcinoma. Cancer Med. 2018, 7, 869–882.
- Li, Z.; Zhang, J.; Liu, X.; Li, S.; Wang, Q.; Di, C.; Hu, Z.; Yu, T.; Ding, J.; Li, J.; et al. The LINC01138 drives malignancies via activating arginine methyltransferase 5 in hepatocellular carcinoma. Nat. Commun. 2018, 9, 1572.
- Zhu, K.; Peng, Y.; Hu, J.; Zhan, H.; Yang, L.; Gao, Q.; Jia, H.; Luo, R.; Dai, Z.; Tang, Z.; et al. Metadherin-PRMT5 complex enhances the metastasis of hepatocellular carcinoma through the WNT-beta-catenin signaling pathway. Carcinogenesis 2020, 41, 130–138.
- Hing, Z.A.; Walker, J.S.; Whipp, E.C.; Brinton, L.; Cannon, M.; Zhang, P.; Sher, S.; Cempre, C.B.; Brown, F.; Smith, P.L.; et al. Dysregulation of PRMT5 in chronic lymphocytic leukemia promotes progression with high risk of Richter’s transformation. Nat. Commun. 2023, 14, 97.
- Qin, Y.; Hu, Q.; Xu, J.; Ji, S.; Dai, W.; Liu, W.; Xu, W.; Sun, Q.; Zhang, Z.; Ni, Q.; et al. PRMT5 enhances tumorigenicity and glycolysis in pancreatic cancer via the FBW7/cMyc axis. Cell Commun. Signal. 2019, 17, 30.
- Orben, F.; Lankes, K.; Schneeweis, C.; Hassan, Z.; Jakubowsky, H.; Krauss, L.; Boniolo, F.; Schneider, C.; Schafer, A.; Murr, J.; et al. Epigenetic drug screening defines a PRMT5 inhibitor-sensitive pancreatic cancer subtype. JCI Insight 2022, 7, e151353.
- Dong, Y.; Song, C.; Wang, Y.; Lei, Z.; Xu, F.; Guan, H.; Chen, A.; Li, F. Inhibition of PRMT5 suppresses osteoclast differentiation and partially protects against ovariectomy-induced bone loss through downregulation of CXCL10 and RSAD2. Cell Signal. 2017, 34, 55–65.
- Kota, S.K.; Roening, C.; Patel, N.; Kota, S.B.; Baron, R. PRMT5 inhibition promotes osteogenic differentiation of mesenchymal stromal cells and represses basal interferon stimulated gene expression. Bone 2018, 117, 37–46.
- Mounir, Z.; Korn, J.M.; Westerling, T.; Lin, F.; Kirby, C.A.; Schirle, M.; McAllister, G.; Hoffman, G.; Ramadan, N.; Hartung, A.; et al. ERG signaling in prostate cancer is driven through PRMT5-dependent methylation of the Androgen Receptor. eLife 2016, 5, e13964.
- Beketova, E.; Fang, S.; Owens, J.L.; Liu, S.; Chen, X.; Zhang, Q.; Asberry, A.M.; Deng, X.; Malola, J.; Huang, J.; et al. Protein Arginine Methyltransferase 5 Promotes pICln-Dependent Androgen Receptor Transcription in Castration-Resistant Prostate Cancer. Cancer Res. 2020, 80, 4904–4917.
- Bao, X.; Zhao, S.; Liu, T.; Liu, Y.; Liu, Y.; Yang, X. Overexpression of PRMT5 promotes tumor cell growth and is associated with poor disease prognosis in epithelial ovarian cancer. J. Histochem. Cytochem. 2013, 61, 206–217.
- Wang, Z.; Kong, J.; Wu, Y.; Zhang, J.; Wang, T.; Li, N.; Fan, J.; Wang, H.; Zhang, J.; Ling, R. PRMT5 determines the sensitivity to chemotherapeutics by governing stemness in breast cancer. Breast Cancer Res. Treat. 2018, 168, 531–542.
- Chiang, K.; Zielinska, A.E.; Shaaban, A.M.; Sanchez-Bailon, M.P.; Jarrold, J.; Clarke, T.L.; Zhang, J.; Francis, A.; Jones, L.J.; Smith, S.; et al. PRMT5 Is a Critical Regulator of Breast Cancer Stem Cell Function via Histone Methylation and FOXP1 Expression. Cell Rep. 2017, 21, 3498–3513.
- Yin, S.; Liu, L.; Brobbey, C.; Palanisamy, V.; Ball, L.E.; Olsen, S.K.; Ostrowski, M.C.; Gan, W. PRMT5-mediated arginine methylation activates AKT kinase to govern tumorigenesis. Nat. Commun. 2021, 12, 3444.
- Hu, D.; Gur, M.; Zhou, Z.; Gamper, A.; Hung, M.C.; Fujita, N.; Lan, L.; Bahar, I.; Wan, Y. Interplay between arginine methylation and ubiquitylation regulates KLF4-mediated genome stability and carcinogenesis. Nat. Commun. 2015, 6, 8419.
- Rengasamy, M.; Zhang, F.; Vashisht, A.; Song, W.M.; Aguilo, F.; Sun, Y.; Li, S.; Zhang, W.; Zhang, B.; Wohlschlegel, J.A.; et al. The PRMT5/WDR77 complex regulates alternative splicing through ZNF326 in breast cancer. Nucleic Acids Res. 2017, 45, 11106–11120.
- Powers, M.A.; Fay, M.M.; Factor, R.E.; Welm, A.L.; Ullman, K.S. Protein arginine methyltransferase 5 accelerates tumor growth by arginine methylation of the tumor suppressor programmed cell death 4. Cancer Res. 2011, 71, 5579–5587.
- Yi, J.; Wu, J. Epigenetic regulation in medulloblastoma. Mol. Cell Neurosci. 2018, 87, 65–76.
- Roussel, M.F.; Stripay, J.L. Epigenetic Drivers in Pediatric Medulloblastoma. Cerebellum 2018, 17, 28–36.
- Dubuc, A.M.; Remke, M.; Korshunov, A.; Northcott, P.A.; Zhan, S.H.; Mendez-Lago, M.; Kool, M.; Jones, D.T.; Unterberger, A.; Morrissy, A.S.; et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 2013, 125, 373–384.
- Alimova, I.; Venkataraman, S.; Harris, P.; Marquez, V.E.; Northcott, P.A.; Dubuc, A.; Taylor, M.D.; Foreman, N.K.; Vibhakar, R. Targeting the enhancer of zeste homologue 2 in medulloblastoma. Int. J. Cancer 2012, 131, 1800–1809.
- Favia, A.; Salvatori, L.; Nanni, S.; Iwamoto-Stohl, L.K.; Valente, S.; Mai, A.; Scagnoli, F.; Fontanella, R.A.; Totta, P.; Nasi, S.; et al. The Protein Arginine Methyltransferases 1 and 5 affect Myc properties in glioblastoma stem cells. Sci. Rep. 2019, 9, 15925.
- Chittka, A.; Nitarska, J.; Grazini, U.; Richardson, W.D. Transcription factor positive regulatory domain 4 (PRDM4) recruits protein arginine methyltransferase 5 (PRMT5) to mediate histone arginine methylation and control neural stem cell proliferation and differentiation. J. Biol. Chem. 2012, 287, 42995–43006.
- Zhou, Z.; Feng, Z.; Hu, D.; Yang, P.; Gur, M.; Bahar, I.; Cristofanilli, M.; Gradishar, W.J.; Xie, X.Q.; Wan, Y. A novel small-molecule antagonizes PRMT5-mediated KLF4 methylation for targeted therapy. EBioMedicine 2019, 44, 98–111.
- Wu, Q.; Schapira, M.; Arrowsmith, C.H.; Barsyte-Lovejoy, D. Protein arginine methylation: From enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discov. 2021, 20, 509–530.
- Feustel, K.; Falchook, G.S. Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors in Oncology Clinical Trials: A review. J. Immunother. Precis. Oncol. 2022, 5, 58–67.
- Millar, H.J.; Brehmer, D.; Verhulst, T.; Haddish-Berhane, N.; Greway, T.; Gaffney, D.; Boeckx, A.; Heerde, E.V.; Nys, T.; Portale, J.; et al. In vivo efficacy and pharmacodynamic modulation of JNJ-64619178, a selective PRMT5 inhibitor, in human lung and hematologic preclinical models . In Proceedings of the American Association for Cancer Research Annual Meeting 2019, Atlanta, GA, USA, 29 March–3 April 2019; AACR: Philadelphia, PA, USA, 2019. Volume 79 (Suppl 13), Abstract nr 950.
- Jensen-Pergakes, K.; Tatlock, J.; Maegley, K.A.; McAlpine, I.J.; McTigue, M.; Xie, T.; Dillon, C.P.; Wang, Y.; Yamazaki, S.; Spiegel, N.; et al. SAM-Competitive PRMT5 Inhibitor PF-06939999 Demonstrates Antitumor Activity in Splicing Dysregulated NSCLC with Decreased Liability of Drug Resistance. Mol. Cancer Ther. 2022, 21, 3–15.
- Duncan, K.W.; Rioux, N.; Boriack-Sjodin, P.A.; Munchhof, M.J.; Reiter, L.A.; Majer, C.R.; Jin, L.; Johnston, L.D.; Chan-Penebre, E.; Kuplast, K.G.; et al. Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666. ACS Med. Chem. Lett. 2016, 7, 162–166.
- Gulla, A.; Hideshima, T.; Bianchi, G.; Fulciniti, M.; Kemal Samur, M.; Qi, J.; Tai, Y.T.; Harada, T.; Morelli, E.; Amodio, N.; et al. Protein arginine methyltransferase 5 has prognostic relevance and is a druggable target in multiple myeloma. Leukemia 2018, 32, 996–1002.
- Hu, G.; Wang, X.; Han, Y.; Wang, P. Protein arginine methyltransferase 5 promotes bladder cancer growth through inhibiting NF-kB dependent apoptosis. EXCLI J. 2018, 17, 1157–1166.
- Kaushik, S.; Liu, F.; Veazey, K.J.; Gao, G.; Das, P.; Neves, L.F.; Lin, K.; Zhong, Y.; Lu, Y.; Giuliani, V.; et al. Genetic deletion or small-molecule inhibition of the arginine methyltransferase PRMT5 exhibit anti-tumoral activity in mouse models of MLL-rearranged AML. Leukemia 2018, 32, 499–509.
- Serio, J.; Ropa, J.; Chen, W.; Mysliwski, M.; Saha, N.; Chen, L.; Wang, J.; Miao, H.; Cierpicki, T.; Grembecka, J.; et al. The PAF complex regulation of Prmt5 facilitates the progression and maintenance of MLL fusion leukemia. Oncogene 2018, 37, 450–460.
- AbuHammad, S.; Cullinane, C.; Martin, C.; Bacolas, Z.; Ward, T.; Chen, H.; Slater, A.; Ardley, K.; Kirby, L.; Chan, K.T.; et al. Regulation of PRMT5-MDM4 axis is critical in the response to CDK4/6 inhibitors in melanoma. Proc. Natl. Acad. Sci. USA 2019, 116, 17990–18000.
- Gerhart, S.V.; Kellner, W.A.; Thompson, C.; Pappalardi, M.B.; Zhang, X.P.; Montes de Oca, R.; Penebre, E.; Duncan, K.; Boriack-Sjodin, A.; Le, B.; et al. Activation of the p53-MDM4 regulatory axis defines the anti-tumour response to PRMT5 inhibition through its role in regulating cellular splicing. Sci. Rep. 2018, 8, 9711.
- Vinet, M.; Suresh, S.; Maire, V.; Monchecourt, C.; Nemati, F.; Lesage, L.; Pierre, F.; Ye, M.; Lescure, A.; Brisson, A.; et al. Protein arginine methyltransferase 5: A novel therapeutic target for triple-negative breast cancers. Cancer Med. 2019, 8, 2414–2428.
- Zhu, F.; Guo, H.; Bates, P.D.; Zhang, S.; Zhang, H.; Nomie, K.J.; Li, Y.; Lu, L.; Seibold, K.R.; Wang, F.; et al. PRMT5 is upregulated by B-cell receptor signaling and forms a positive-feedback loop with PI3K/AKT in lymphoma cells. Leukemia 2019, 33, 2898–2911.
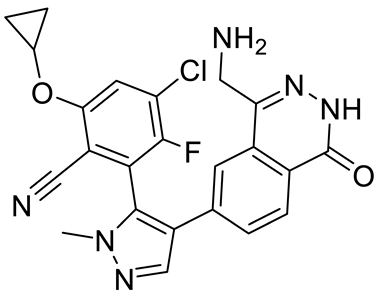
- Villalona-Calero, M.A.; Patnaik, A.; Maki, R.G.; O’Neil, B.; Abbruzzese, J.L.; Dagogo-Jack, I.; Devarakonda, S.; Wahlroos, S.; Lin, C.-C.; Fujiwara, Y.; et al. Design and rationale of a phase 1 dose-escalation study of AMG 193, a methylthioadenosine (MTA)-cooperative PRMT5 inhibitor, in patients with advanced methylthioadenosine phosphorylase (MTAP)-null solid tumors. J. Clin. Oncol. 2022, 40, TPS3167.
- Bhagwat, N.; Zhang, Y.; Lin, H.; Wang, M.; Rominger, D.; Emm, T.; Chugani-Mahtani, D.; Angelis, D.; Shetty, R.; Leal, R.; et al. Preclinical Characterization of PRT543, a Potent and Selective Inhibitor of Protein Arginine Methyltransferase 5 (PRMT5), with Broad Antitumor Activity in In vitro and In vivo Models. In Proceedings of the Annual Meeting of the American Association for Cancer Research, Philadelphia, PA, USA, 27–28 April 2020.
- Falchook, G. A phase 1 dose-escalation study of protein arginine methyltransferase 5 (PRMT5) brain-penetrant inhibitor PRT811 in patients with advanced solid tumors, including recurrent high-grade gliomas. In Proceedings of the AACR-NCI-EORTC Virtual Conference, Virtual, 7–10 October 2021; Available online: www.aacr.org/meeting/aacr-nci-eortc-international (accessed on 17 November 2023).
- Zhang, M.; Tsai, A.; Cottrell, K.M.; Haines, B.B.; Wilker, E.; DiBenedetto, H.; Weitzman, R.; Huang, A.; Davis, C.B.; Maxwell, J.P.; et al. TNG908, a brain-penetrant MTA-cooperative PRMT5 inhibitor, is efficacious in preclinical glioblastoma models. In Proceedings of the American Association for Cancer Research Annual Meeting 2023, Orlando, FL, USA, 14–19 April 2023; Part 1 (Regular and Invited Abstracts). AACR: Philadelphia, PA, USA, 2023; Volume 83.
- Vegar, L.; Aranda, R.; Waters, L.; Moya, K.; Hebbert, A.; Smith, C.S.; Hallin, J.; David, B.M.; Engstrom, L.D.; Vanderpool, D.; et al. A novel MTA-cooperative PRMT5 inhibitor, MRTX1719, stabilizes the ternary MTA-PRMT5 complex and leads to synthetic lethality in MTAP deleted cancers . In Proceedings of the American Association for Cancer Research Annual Meeting 2023, Orlando, FL, USA, 14–19 April 2023; Part 1 (Regular and Invited Abstracts). AACR: Philadelphia, PA, USA, 2023; Volume 83.
- Engstrom, L.D.; Aranda, R.; Waters, L.; Moya, K.; Bowcut, V.; Vegar, L.; Trinh, D.; Hebbert, A.; Smith, C.R.; Kulyk, S.; et al. MRTX1719 is an MTA-cooperative PRMT5 inhibitor that exhibits synthetic lethality in preclinical models and patients with MTAP deleted cancer. Cancer Discov. 2023, 13, 2412–2431.
- Bonday, Z.Q.; Cortez, G.S.; Grogan, M.J.; Antonysamy, S.; Weichert, K.; Bocchinfuso, W.P.; Li, F.; Kennedy, S.; Li, B.; Mader, M.M.; et al. LLY-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, with Antitumor Activity. ACS Med. Chem. Lett. 2018, 9, 612–617.
- Snyder, K.J.; Zitzer, N.C.; Gao, Y.; Choe, H.K.; Sell, N.E.; Neidemire-Colley, L.; Ignaci, A.; Kale, C.; Devine, R.D.; Abad, M.G.; et al. PRMT5 regulates T cell interferon response and is a target for acute graft-versus-host disease. JCI Insight 2020, 5, e131099.
- Palte, R.L.; Schneider, S.E.; Altman, M.D.; Hayes, R.P.; Kawamura, S.; Lacey, B.M.; Mansueto, M.S.; Reutershan, M.; Siliphaivanh, P.; Sondey, C.; et al. Allosteric Modulation of Protein Arginine Methyltransferase 5 (PRMT5). ACS Med. Chem. Lett. 2020, 11, 1688–1693.
- Sivanandhan, D.; Gajendran, C.; Mohammed, Z.; Gosu, R.; Sher, D.; Mansur, S.; Friedmann-Morvinski, D.; Rajagopal, S.; Rastelli, L. JBI-778, a novel brain-penetrant small molecule PRMT5 inhibitor for treatment of cancer . In Proceedings of the American Association for Cancer Research Annual Meeting 2023, Orlando, FL, USA, 14–19 April 2023; Part 1 (Regular and Invited Abstracts). AACR: Philadelphia, PA, USA, 2023; Volume 83.
- Zheng, J.; Li, B.; Wu, Y.; Wu, X.; Wang, Y. Targeting Arginine Methyltransferase PRMT5 for Cancer Therapy: Updated Progress and Novel Strategies. J. Med. Chem. 2023, 66, 8407–8427.