Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Abdalla Abdrabou and Version 2 by Rita Xu.
IκB kinases (IKKs), specifically IKKα and IKKβ, have long been recognized for their pivotal role in the NF-κB pathway, orchestrating immune and inflammatory responses.
- IκB kinases
- IKKs
- NF-κB
1. Introduction
The NF-κB (Nuclear Factor-κB) pathway is a linchpin of cellular responses to external stimuli, especially in the realms of immune and inflammatory processes [1]. Within the broader context of the NF-κB signaling pathway, the IκB kinases (IKKs), particularly IKKα and IKKβ, occupy a pivotal position in the intricate regulatory network of these pathways. Traditionally recognized for their roles in immune surveillance and defense, the IKKs have recently emerged as enigmatic figures in the landscape of cancer biology [2][3][4][5][2,3,4,5]. Their Janus-faced nature, promoting or suppressing tumorigenesis depending on context, has prompted an intensive exploration of their molecular mechanisms within the context of cancer and the possibility of targeting them [6].
The NF-κB pathway, with its intricate family members, represents a dynamic signaling network that orchestrates cellular responses to a multitude of extracellular signals [7][13]. The fundamental aspect of this pathway revolves around the NF-κB transcription factors, comprising various members such as p65 (RelA), RelB, c-Rel, p105/p50, and p100/p52. These members of the NF-κB family combine in different dimeric forms, each playing specific roles in overseeing signaling pathways, particularly those involved in immune responses [8][9][10][14,15,16].
Central to the regulation of NF-κB is the presence of inhibitory proteins referred to as IκBs, responsible for maintaining NF-κB dimers in an inactive state within the cell’s cytoplasm [11][17]. The activation of the pathway involves the phosphorylation of IκBs, tagging them for degradation by the proteasome machinery. This process liberates NF-κB dimers, allowing them to migrate into the nucleus and commence gene transcription (Figure 1) [12][18].
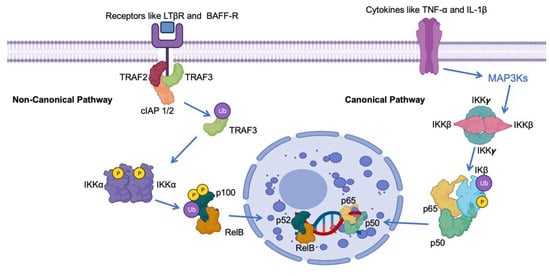
Figure 1. Schematic representation of the roles of IκB in the canonical and non-canonical NF-κB pathway. The NF-kB pathway involves key proteins such as Tumor Necrosis Factor Receptor-Associated Factor 3 (TRAF3), Precursor protein 100 (p100), V-rel avian reticuloendotheliosis viral oncogene homolog B (RelB), Nuclear Factor NF-kappa-B p52 subunit (p52), Nuclear Factor NF-kappa-B p65 subunit (p65), Cellular Inhibitor of Apoptosis Protein 1/2 (cIAP 1/2), B-cell Activating Factor Receptor (BAFF-R), and Lymphotoxin Beta Receptor (LTβR). Ub indicates ubiquitination, IKK α ((IkappaB kinase alpha). P indicates phosphorylation.
The NF-κB pathway, essential for various physiological processes, exists in multiple branches. Primarily, there are two well-characterized pathways: the canonical (or classical) and non-canonical (or alternative) [13][19]. These pathways differ in terms of the stimuli that activate them, the proteins involved, and the nature of their functions. The effective enhancement of NF-κB involves a complex regulation process mediated by the IκB kinase (IKK) complex. This intricate system orchestrates the phosphorylation of IκB proteins, leading to their ubiquitination and subsequent degradation via the proteasome [14][15][16][20,21,22]. This series of events ultimately results in the liberated NF-κB complexes translocating into the nucleus.
Within the nucleus, these NF-κB complexes engage with specific DNA sequences, thereby governing the transcription of genes involved in diverse processes, including immune responses, cellular growth regulation, and the modulation of cell survival [17][18][19][23,24,25]. Notably, within the context of cancer, NF-κB-dependent genes encompass those responsible for encoding cytokines, chemokines, cyclin D1, matrix metalloproteinases, and antiapoptotic proteins such as Bcl-xL [20][21][22][26,27,28].
2. Canonical NF-κB Pathway
In the Canonical Nuclear Factor-kappa B (NF-κB) pathway, activation is initiated by a diverse array of stimuli, encompassing proinflammatory cytokines like Tumor Necrosis Factor-alpha (TNF-α) and Interleukin-1 beta (IL-1β), as well as microbial products like lipopolysaccharides (LPSs) (Figure 1) [23][24][25][26][29,30,31,32]. Key to this pathway is the activation of IKKβ, which subsequently phosphorylates and targets IκBα and IκBβ for degradation. As a result, this liberation allows for the movement of p50-RelA dimers into the nucleus. Once in the nucleus, these dimers function as transcription factors, overseeing the expression of genes linked to inflammation, immune responses, and cell survival (Figure 1) [27][28][29][33,34,35].
3. Non-Canonical NF-κB Pathway
Conversely, the Non-Canonical NF-κB pathway is typically triggered by a unique group of receptors, which encompass the lymphotoxin-β receptor (LTβR) and B-cell activating factor receptor (BAFF-R) [30][36]. This pathway is reliant on the conversion of p100 to p52, a process orchestrated by the activation of IKKα. Subsequently, the p52-RelB dimers relocate to the nucleus, assuming critical roles in the development of secondary lymphoid organs, B-cell maturation, and the organization of lymphoid tissues (Figure 1) [31][32][33][37,38,39].
NIK, or NF-κB-inducing kinase, holds a crucial position in regulating the non-canonical NF-κB signaling pathway [34][40]. Initially acknowledged for its role in activating the canonical NF-κB pathway, the absence of NIK did not hinder the TNF-induced IKKβ/p65/p50 activation. However, it was later discovered to be essential for triggering the non-canonical NF-κB pathway [35][36][37][41,42,43]. The regulation of NIK predominantly occurs post-translationally. Structurally, NIK encompasses four domains: a TRAF3-binding N-terminal region, a negative regulatory domain (NRD), a core kinase domain, and a C-terminal domain responsible for binding with proteins like IKKα and p100 [38][39][40][44,45,46].
Initially recognized as a mediator following TNF and IL-1 receptor activation, NIK’s kinase activity was deemed crucial in facilitating this particular process. Additionally, it mediates stimulation through various receptors like CD27, CD30, CD40, LTβR, and BAFFR [41][42][43][44][47,48,49,50]. The overexpression of NIK activates NF-κB, protecting cells from TNF-induced apoptosis, while kinase-dead NIK mutants inhibit NF-κB activation by TNFα [45][46][47][51,52,53].
Under normal conditions, NIK binds to TRAF2/3 and cIAP1/2, leading to its continuous ubiquitination and degradation [48][54]. Stimulation by cytokines (such as CD40L, TWEAK, LTα/β, or LPS) sequesters TRAF2/3, allowing the cIAP1-mediated ubiquitination of TRAF3 [49][55]. The subsequent degradation of TRAF3 leads to the accumulation of newly synthesized NIK within the cell. This stabilization and buildup of NIK are crucial for initiating the noncanonical NF-κB pathway [50][51][52][56,57,58]. Upon receptor activation, NIK triggers IKKα phosphorylation at Ser-176 and Ser-180, activating it to phosphorylate p100. The phosphorylation of p100 prompts the binding to ubiquitin ligase β-TrCP, resulting in partial proteasomal processing to p52. This processing removes the inhibitory C-terminal ankyrin repeat domain of p100, akin to the function of mature IκB proteins, thus maintaining RelB inactive in the cytoplasm. Subsequently, p52-RelB translocates to the nucleus to regulate transcription [53][54][55][56][59,60,61,62].
NIK interacts with and activates both IKKα and IKKβ, phosphorylating IKKα to a greater extent. Consequently, NIK acts as an upstream kinase for the IKK complex, facilitating signaling from multiple cytokine receptors [57][58][59][63,64,65]. Several other kinases, including MEKK1 and TAK1, were identified as IKK kinases, sometimes acting alongside NIK. TAK1, for instance, can activate NIK/IKK/NF-κB signaling independently of NIK in certain contexts [60][66]. Additionally, proteins like TRAF2, 5, and 6, as well as TBK-1, contribute to NF-κB activation by acting upstream of NIK. Moreover, Bcl10 has been reported to phosphorylate NIK under specific inflammatory conditions in human colonic epithelial cells treated with carrageenan (CGN) [16][61][62][22,67,68].
4. The Dark Side: IκB Kinases as Tumor Promoters
4.1. IKKα (Inhibitor of κB Kinase Alpha)
IKKα plays a multifaceted role in cancer, impacting both its initiation and progression, along with metastasis. In colorectal cancer cells (HT29), IKKα exhibits abnormal activation within the nucleus of tumor cells [63][69]. Here, it binds to specific genes reliant on Notch signaling, such as hes1 and herp2. The nuclear IKKα phosphorylates a nuclear co-repressor, SMRT, causing its release from chromatin and the subsequent expression of Notch-dependent genes [64][70], leading to more aggressive growth and proliferation. Pan-IKK inhibition re-establishes SMRT chromatin binding, curbing Notch-related gene expression, and restraining tumor growth in experimental models [65][71].
Additionally, IKKα phosphorylates N-CoR, akin to SMRT, facilitating its nuclear export from CRC cells [66][72]. The active nuclear IKKα isoform, IKKα(p45), is crucial for preventing apoptosis and thereby fostering tumor growth, specifically in HCT116 cells. Mechanistically, the association between active TAK1, BRAF, a complex containing IKKα(p45), and NEMO leads to SMRT and Histone H3 phosphorylation, which is vital for BRAF-mediated transformation independent from NF-κB signaling [67][73].
In keratinocytes, evidence demonstrates IKKα’s involvement in cancer initiation independently of NF-κB [68][74]. The deletion of IKKα induces skin squamous cell carcinoma in mice, affecting 14-3-3σ expression and prompting aberrant cell proliferation, disrupting skin homeostasis, and promoting cell transformation [69][75]. Additional studies support IKKα’s tumor suppressor role in the skin, linking its activity to the transforming growth factor beta (TGFβ) pathway. Moreover, a specific variant of nuclear IKKα in keratinocytes leads to more aggressive tumors upon exposure to chemical carcinogens [70][71][76,77].
Basal cell carcinomas (BCCs) are the most prevalent among human cancers affecting the skin [72][78]. While the noncanonical NF-κB pathway relies on IKKα, its specific role in BCC remains unclear. One study indicated that, within both BCC and non-malignant conditions, IKKα is present in the nucleus. Within BCC, the nuclear IKKα directly interacts with the promoters of inflammation factors and LGR5, a marker for stem cells. This interaction leads to an increase in LGR5 expression through the activation of the STAT3 signaling pathway, thereby contributing to cancer progression. The activation of the STAT3 pathway influences the LGR5 expression in a manner dependent on IKKα, as demonstrated by the interplay between STAT3 and IKKα. Moreover, suppressing the IKKα impedes the tumor growth and transition from the epithelial stage to the mesenchymal stage. This finding highlights IKKα’s role as a genuine chromatin regulator in BCC. Its heightened expression facilitates oncogenic transformation by promoting the expression of genes related to stemness and inflammation. Consequently, these findings offer a fresh perspective on how IKKα may participate in the progression of BCC tumors within an inflammatory microenvironment [73][79].
Moreover, in a study by Mahato and colleagues [74][80], they have shown that the suppression of IKKα in prostate cancer cells using synthetic siRNAs affects tumor cell growth and invasiveness. TIn this study, the authors designed three synthetic siRNAs targeting the specific regions of IKKα mRNA and evaluated their ability to silence IKKα in PC-3 and DU145 cells. A range of assays, including wound healing, migration, proliferation, and cell cycle analysis, were employed to investigate how IKKα siRNAs biologically impacted prostate cancer cells. Interestingly, their results uncovered potent siRNAs that could silence IKKα by up to 70%, resulting in decreased wound healing, migration, invasion, and cell attachment capabilities in prostate cancer cells. Additionally, this study observed comparable anti-invasive effects in the presence of RANKL. However, silencing IKKα had minimal effects on cell proliferation and cell cycle distribution. These findings strongly indicate that IKKα significantly influences prostate cancer invasion and metastasis while playing a minor role in cell proliferation. Targeting IKKα using siRNA emerges as a promising therapeutic approach for managing prostate cancer by reducing invasion and metastasis without directly impacting cell proliferation [75][76][81,82].
Moreover, IKKα contributes to progesterone-induced tumor promotion in breast cancer, downstream of RANKL induction, and fosters metastatic spread relying on RANKL produced by tumor-infiltrating regulatory T cells. It phosphorylates Estrogen Receptor α, its coactivator AIB1/SRC3, and induces targets like cyclin D1 and c-myc, driving breast cancer cell proliferation [77][83]. Clinical observations link IKKα expression in breast cancer cells with patient outcomes regardless of cellular localization. In triple-negative breast cancer (TNBC) cells, IKKα mediates Notch signaling triggered by the Notch ligand Jagged1, a pivotal pathway for TNBC Cancer Stem Cell survival [78][84]. Combining therapies targeting the intersection of Notch, AKT, and NF-κB pathways holds promise for therapeutic applications against cancer stem cells in TNBC [79][85].
4.2. IKKβ (Inhibitor of κB Kinase Beta)
In contrast to IKKα, IKKβ is predominantly associated with the canonical NF-κB pathway. One of its key functions is the phosphorylation of IκBα and IκBβ. This phosphorylation event marks IκB proteins for degradation, allowing the release of p50-RelA dimers [80][86]. The degradation of IκB proteins leads to the liberation of NF-κB, a transcription factor crucial in orchestrating the expression of pro-inflammatory genes. This includes genes responsible for cytokine and chemokine production, thus fostering a sustained and amplified inflammatory response within the tumor microenvironment [81][87]. This persistent inflammation, driven by IKKβ, creates a milieu that nurtures tumor growth, angiogenesis, and metastasis [82][88].
The released dimers subsequently move into the nucleus, where they commence the transcription of genes linked with the canonical NF-κB pathway. Notably, the activation of IKKβ is frequently prompted by proinflammatory stimuli, connecting it to persistent inflammation [83][89]. This linkage further underscores IKKβ’s significance in fostering tumor progression, the formation of new blood vessels (angiogenesis), and the spread of cancer to distant sites (metastasis), highlighting its critical role in the context of cancer. However, it is important to note that the outcome of this inflammatory response is context-dependent, either influencing the promotion of tumor formation or the initiation of an immune reaction against tumors [84][90].