Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Luisa Maia and Version 3 by Lindsay Dong.
Living organisms use selenium mainly in the form of selenocysteine in the active site of oxidoreductases. Here, selenium’s unique chemistry is believed to modulate the reaction mechanism and enhance the catalytic efficiency of specific enzymes in ways not achievable with a sulfur-containing cysteine. However, despite the fact that selenium/sulfur have different physicochemical properties, several selenoproteins have fully functional cysteine-containing homologues and some organisms do not use selenocysteine at all.
- selenium
- biology
1. Introduction
Discovered in 1817, selenium was for long regarded as a toxic element [1][2][3][1,2,3] and only in the second half of the XX century was it demonstrated to be essential for all forms of life [4][5][6][7][8][9][10][4,5,6,7,8,9,10]. Living organisms have learned to harness the unique chemical features provided by selenium (over sulfur) and use this element mainly in the active site of oxidoreductases in the form of selenocysteine, an amino acid genetically encoded by a specific codon (UGA) that is considered the 21st amino acid.
Several selenocysteine-containing enzymes evolved to play essential roles in various biological processes. Still, some of those selenoproteins have fully functional cysteine-containing homologues and some organisms do not use selenocysteine at all. Hence, understanding the biological use of selenium is of considerable interest.
2. Selenium versus Sulfur
Selenium is a chemical element belonging to the chalcogens family of the Periodic Table (Group 16). It resembles the “lighter” sulfur in some chemical features and, in Biology, selenium can be found replacing sulfur in two amino acids: selenocysteine (Se-Cys) and selenomethionine (Se-Met). However, in spite of the similarities, many significant chemical differences exist between these two chalcogens [11][12][13][14][15][11,12,13,14,15]. As sulfur, selenium can display a wide range of oxidation states (from −2 to +6), but its preference for lower oxidation states and higher reactivity sets it apart from sulfur. Its reactions are often also faster than its sulfur counterparts because selenium is more polarizable (softer). Its larger spin—orbit coupling (compared to sulfur) probably facilitates spin-forbidden reactions, as the ones involved in the rapid oxidation of selenocysteine under air (compared to cysteine oxidation). Moreover, the selenocysteine selenol’s lower pKa value (5.2, compared to 8.3 of cysteine thiol) is expected to favor its deprotonation and nucleophilic character at physiological pH [16], while the weaker Se-H bond makes the selenocysteine less basic, compared to cysteine [17][18][17,18]. The biologically relevant redox chemistry is also significantly different in these two elements [19][20][21][19,20,21]. The selenocysteine one-electron oxidation-derived radical is more easily formed ((RSe•/RSeH) = 0.43 V versus (RS•/RSH) = 0.92 V [22]) and relatively more stable than the cisteine radical [22][23][24][22,23,24]. As a result, for example, while the cysteine radical can oxidize a tyrosine residue (to yield tyrosine radical), the selenocysteine radical can not [22]. Also noteworthy are the thiol/disulfide exchange reactions, where the selenocysteine reactions (Se-Cys/Cys-Se-Se-Cys) are faster than the cysteine ones [12][14][25][26][12,14,25,26].3. Formate Dehydrogenase
FDH was one of the first enzymes demonstrated to contain selenium and a selenocysteine-specific codon (TGA) in its gene sequence (Clostridium thermoaceticum and E. coli enzymes) [27][28][27,28]. Those seminal works were essential to overcome the prevailing idea that selenium was (only) a toxic substance and lead to its recognition as an essential element (also for mammals and humans by contemporary works). In spite of being one of the most widely distributed selenoproteins (probably due to its extensive lateral gene transfer, together with the corresponding selenocysteine synthesis and incorporation system) [29], FDH constitutes a key example where, as far as is presently known, selenium does not present any clear advantage over sulfur. Contrary to other selenoenzymes, living organisms hold both active selenocysteine- and cysteine-containing FDH homologues and, thus, the selenium role in FDH catalysis remains, so far, elusive.3.1. The Current Picture
3.1.1. Enzymatic Machinery
FDHs catalyze the two-electron interconversion of formate and carbon dioxide (Equation (1)) in diverse metabolic pathways, operating in different subcellular locations, such as C1 metabolism, carbon dioxide fixation (carbon assimilation), and to derive energy (coupling formate oxidation to the reduction of different terminal electron acceptors) [30][31][32][33][34][35][36][37][38][30,31,32,33,34,35,36,37,38]. Since each pathway requires a specific “FDH enzymatic machinery” to accomplish the respective biological function, FDHs evolved as a highly heterogeneous group of enzymes, displaying diverse structural (subunits) organization and composition of redox-active centers [39][40][41][42][43][44][45][46][47][48][39,40,41,42,43,44,45,46,47,48]. HCOO−
⇌ CO2
+ 2e−
+ H+
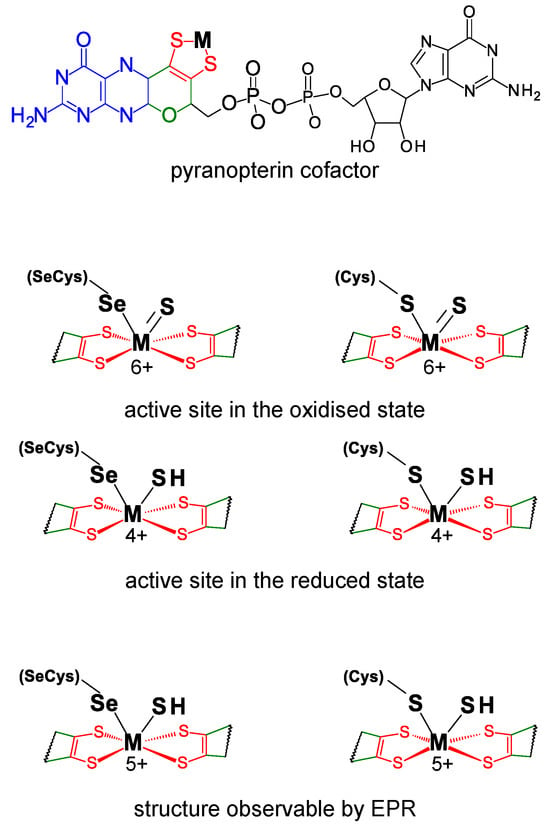
FDHs can be divided into two main classes. The metal-independent FDH class comprises enzymes, typically homodimers that have no metal ions or other redox-active centers, nor selenium [49][50][51][52][53][54][49,50,51,52,53,54]. These enzymes, found in bacteria, fungi, and plants, are NAD-dependent and belong to the D-specific dehydrogenases of the 2-oxyacids family. On the contrary, the metal-dependent FDH class, present only in prokaryotes, comprises enzymes that harbor different redox-active centers and display high structural diversity [41][42][43][45][46][48][41,42,43,45,46,48]. As the class name indicates, the active site of these enzymes holds one molybdenum or one tungsten ion in a very well conserved metal center (Figure 1). In its oxidized (6+) form, the metal (molybdenum or tungsten) is coordinated by the cis-dithiolene (–S–C=C–S–) group of two pyranopterin cofactor molecules, one terminal sulfido group (Mo6+/W6+=S), plus one selenium or one sulfur atom from a selenocysteine or cysteine residue (Mo6+/W6+-Se(Cys) or Mo6+/W6+-S(Cys)) (abbreviated as SeCys-Mo-FDH, SeCys-W-FDH, Cys-Mo-FDH, and Cys-W-FDH) [40][44][55][56][40,44,55,56]. Noteworthy, there is no apparent relation (as far as is presently known) between the metal (molybdenum or tungsten) and the presence of a selenocysteine or cysteine residue and catalytically efficient SeCys-Mo-FDH, SeCys-W-FDH, Cys-Mo-FDH, and Cys-W-FDH have been known for a long time.
Figure 1. Active site structure of metal-dependent FDHs and FMFDHs. Top: Structure of the pyranopterin cofactor. The pyranopterin cofactor molecule is formed by pyrano(green)–pterin(blue)–dithiolene(red)–methylphosphate(black) moieties; in all so far characterized enzymes, the cofactor is found esterified with a guanosine monophosphate (dark gray). The dithiolene (–S–C=C–S–) group forms a five-membered ene-1,2-dithiolene chelate ring with the molybdenum or tungsten ion, here indicated as M (from metal). Middle: Structure of the active site in the oxidized and reduced state. Bottom: Active site structure supported by EPR data. In middle and bottom structures, for simplicity, only the dithiolene moiety of the pyranopterin cofactor is represented.
Similar to FDHs, selenocysteine-containing and cysteine-containing N-formyl-methanofuran dehydrogenases (SeCys-FMFDH and Cys-FMFDH) exist and selenium’s role in FMFDH catalysis is unknown as well. FMFDHs are FDH-like enzymes that have two physically separated active sites: one catalyzes the reduction of carbon dioxide to formate, which is then intramolecularly transferred to the second active site, where it is condensed with methanofuran to form N-formyl-methanofuran [57][58][59][60][57,58,59,60].
3.1.2. Reaction Mechanism
Regardless of the physiological function and structural complexity, the reaction mechanism of the interconversion of formate and carbon dioxide (Equation (1)) is believed to be similar in all these selenocysteine- and cysteine-containing FDH and FMFDH enzymes. As originally proposed by Niks et al. [61] for formate oxidation and shortly after also for carbon dioxide reduction by Maia et al. [62], it is currently well established that formate oxidation and carbon dioxide reduction proceed through hydride transfer, with the oxidized and reduced active site sulfido group, Mo/W6+=S and Mo/W4+-SH, acting as the direct hydride acceptor and donor, respectively (Figure 2) [63][64][65][66][63,64,65,66] (even though other atomic details of the reaction mechanism are not yet consensual; see, for example [67]). It is noteworthy that no direct role in the chemical transformations is presently ascribed to the selenocysteine or cysteine residue, in accordance with the existence of catalytically efficient SeCys enzymes and Cys enzymes (a similar situation occurs with molybdenum and tungsten).
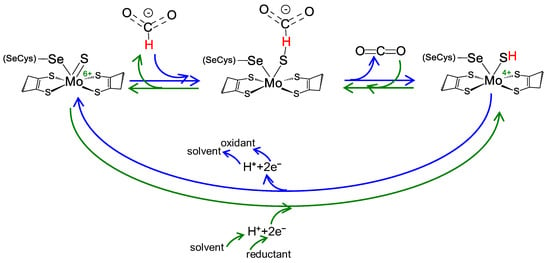
Figure 2. Reversible FDH and FMFDH reaction mechanism, as proposed by Maia et al. [62]. Reaction mechanism proposed for formate oxidation (blue arrows) and carbon dioxide reduction (green arrows) for both metal-dependent FDHs and FMFDHs. For simplicity, the mechanism is represented for a molybdenum, selenocysteine-containing enzyme, but it should be similar for tungsten and cysteine-containing enzymes. See text for details.
Briefly, formate oxidation (Figure 2, blue arrows) is initiated with the formate binding to the oxidized active site but not directly to the molybdenum/tungsten atom. Formate is suggested to bind in a binding pocket, where a conserved arginine residue “anchors” its oxygen atom(s) through hydrogen bond(s), and forces its Cα hydrogen to point towards the sulfido ligand (Mo6+/W6+=S). Subsequently, formate oxidation proceeds by a straightforward hydride transfer from formate to the sulfido group of the oxidized molybdenum/tungsten centre, leading to the formation of Mo/W4+-SH and CO2. The re-oxidation of Mo/W4+ to Mo/W6+ (via intramolecular electron transfer to the enzyme’s other redox center(s) and, eventually, to the physiological partner) and the release of carbon dioxide close the catalytic cycle. The now oxidized Mo/W6+ favors the sulfido group deprotonation (dictated by the ligand pKa [68][69][70][68,69,70]) and the initial oxidized metal centre, Mo/W6+=S, is regenerated. Under non-steady-state catalytic conditions (such as the ones created in EPR experiments described below), the molybdenum/tungsten one-electron oxidation should be favored (Mo/W4+→Mo/W5+), leading to the formation of the EPR detectable species.
The carbon dioxide reduction is suggested to follow the reverse reaction mechanism (Figure 2, green arrows) but starting with a reduced active site, holding a protonated sulfido group, Mo/W6+-SH (as is dictated by the ligands pKa [68][69][70][68,69,70]). Carbon dioxide is suggested to bind to the same binding pocket, where the arginine residue is key to anchor it in the correct position to orient its carbon atom towards the protonated sulfido. Afterwards, the reaction proceeds through straightforward hydride transfer from the protonated sulfido group. This yields a formate moiety and Mo/W6+=S. The subsequent re-reduction of Mo/W6+ to Mo/W4+ (via intramolecular electron transfer from the enzyme’s physiological partner, through its redox center(s)) and formate release closes the catalytic cycle. The now reduced Mo/W4+ favors the sulfido group protonation and the initial reduced molybdenum/tungsten center, Mo/W4+-SH, is regenerated.
3.2. How Was the Selenium Locus in Formate Dehydrogenases Established?
The presence and essentiality of selenium was demonstrated in pioneer works, mainly in the 1970s, following the incorporation in target enzymes of selenium-75 (present in the growth medium/feed). Actually, FDH was among the first enzymes shown to contain selenium [27][28][27,28].
The recognition of the presence of molybdenum or tungsten and selenium led to a series of spectroscopic studies that were decisive to the early characterization of the FDH active site. Electron paramagnetic resonance spectroscopy (EPR) was thoroughly explored (reviewed recently, for example, in [71][72][73][71,72,73]). In fact, the first evidence for the direct binding of selenium to a metal (molybdenum) active site center was obtained precisely with EPR [74][75][74,75]. The E. coli SeCys-Mo-FDH H was one of the first FDHs to be explored [75][76][75,76]. When reduced with formate, it gives rise to a nearly axial Mo5+ signal, with g1 = 2.094 and g2,3 = 2.001, 1.990, that displays coupling to one formate-derived solvent-exchangeable proton (A1,2,3(1H) = 7.5, 18.9, 20.9 MHz). When the EPR signal is generated from the 77Se-enriched enzyme, a very strong and anisotropic interaction is observed (A1,2,3(77Se) = 13.2, 75, 240 MHz [77], values that are almost five times higher than the ones observed in Mo-Se model compounds [77][78][77,78]). This strong interaction, observed simultaneously with the expected 95,97Mo hyperfine coupling, confirmed that the selenocysteine selenium atom is directly coordinated to the molybdenum (Figure 1) and suggested that the unpaired electron is delocalized 17–27% over the selenium (a finding in line with the expected covalency introduced by selenium in a Mo-Se bond) [77].
These original studies with E. coli FDH H were supported and consolidated with other selenium-containing FDHs, including Desulfovibrio desulfuricans [79], D. gigas [80][81][84,85], D. vulgaris Hildenborough [82][83][84][85][80,81,82,83], and Methylosinus trichosporium [86] FDHs. These enzymes display rhombic Mo5+/W5+ EPR signals with small anisotropy, a well-resolved hyperfine structure due to 95,97Mo/183W, and interaction with a solvent-exchangeable proton (for example: D. desulfuricans: g1,2,3 = 2.012, 1.996, 1.985, A1,2,3(solvent-exchangeable 1H) = 23.1, 29.9, 27.8 MHz [79]; D. vulgaris Hildenborough FDH 1: g1,2,3 = 1.995, 1.881, 1.852, A1,2,3(183W)= 225, 129, 134 MHz [82][80]; D. vulgaris Hildenborough FDH 2 (main component): g1,2,3 = 1.982, 1.876, 1.902, A1,2,3(183W)= 232, 119, 151 MHz [84][82]). The D. desulfuricans FDH displays also a hyperfine interaction with a second non-solvent-exchangeable proton (A1 = 35.1 MHz, A2,3 not detectable) that was assigned to the metal-bound selenocysteine Cβ hydrogen atoms [79]. Together, the EPR data suggest an FDH active site holding a stable selenocysteine–metal ligation. It also suggests that the active site holds a transient proton-accepting site (within the metal magnetic contact) that was assigned as the terminal sulfido group (please see Note above) [61][62][61,62].
The SeCys-FDH active site was also explored by X-ray absorption spectroscopy (XAS) since early times [87]. XAS at the molybdenum and selenium K-edges of the most explored model FDH, E. coli SeCys-Mo-FDH H, revealed four Mo-S ligands at 2.35 Å, one (originally not assigned) Mo=S at 2.1 Å, and one Mo-Se ligand at 2.62 Å, in both oxidized and reduced enzyme [88]. In the D. desulfuricans SeCys-Mo-FDH, the molybdenum and selenium K-edges data also showed a hexa-coordinated active site, with one Mo-Se ligand at 2.57 Å in both oxidized and reduced enzyme [89]. It is noteworthy that the replacement of the E. coli SeCys-Mo-FDH H selenocysteine by a cysteine residue abolished the Mo-Se fingerprint and gave rise to a spectrum consistent with five Mo-S ligands and one Mo=O at 1.7 Å [88]. Comparatively, XAS studies of native Cys-FDHs (for example, oxidized Rhodobacter capsulatus Cys-Mo-FDH [90][91][90,91]) confirmed that the cysteine residue is bound to the metal, as expected. Hence, the XAS results are in excellent agreement with the EPR proposed FDH active site structure, Mo5+/W5+-Se(Cys)(-SH) (Figure 1).
The crystallographic structure of different native SeCys- (and Cys-) FDHs entirely supports this active site structure. The first FDH 3D structure solved, in 1997, was the one of the model E. coli SeCys-Mo-FDH H [92] and this was the only one known for 5 years (2002), when the structure of two more enzymes were finally solved, the E. coli SeCys-Mo-FDH N [93] and Desulfovibrio gigas SeCys-W-FDH [94] (Figure 3). The first FMFDH structure (the Methanothermobacter wolfeii Cys-W-FMFDH) was revealed only 14 years after, in 2016 [59]. Presently, several structures are known [60][82][83][95][96][97][98][99][60,80,81,95,96,97,98,99] and the active site structure is firmly established to be the conserved Mo6+-Se(Cys)(=S), W6+-Se(Cys) (=S), Mo6+-S(Cys)(=S), or W6+-S(Cys)(=S) (Figure 1).
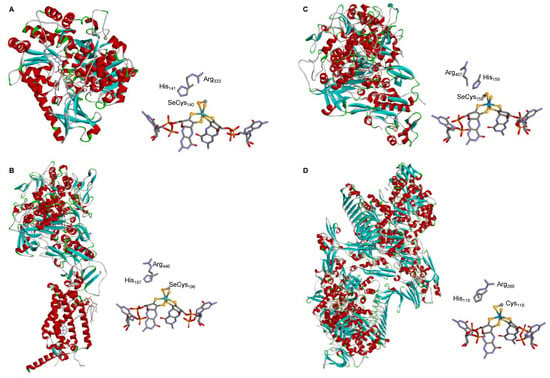
Figure 3. Three-dimensional structure view of some metal-dependent FDHs and FMFDHs and their active sites. (A) E. coli SeCys-Mo-FDH H [92]; (B) E. coli SeCys-Mo-FDH N [93]; (C) D. gigas SeCys-W-FDH [94]; (D) M. wolfeii Cys-W-FMFDH [59]. The structures shown are based on the PDB files 1FDO (A), 1KQF (B), 1H0H (C), and 5T5I (D) (α helices and β sheets are shown in red and cyan, respectively).
3.3. Why Do Some Formate Dehydrogenases Have a Selenocysteine and Not the Less “Expensive” Cysteine Residue?
Since its early identification as a selenium-containing enzyme, the role of selenium in FDH catalysis has intrigued the scientific community. A pioneer work in the late 1980s [100] with the model E. coli SeCys-Mo-FDH H showed that selenocysteine (SeCys140) replacement with a cysteine residue resulted in significant lower FDH activity, while replacement with a serine residue rendered the enzyme inactive. In a subsequent, more comprehensive work by the Stadtman group [101], it was clearly shown that selenocysteine replacement with a cysteine resulted in a marked decrease in FDH activity (kcat/Kmformate (SeCys-FDH) = 108 × 103 M−1s−1 to kcat/Kmformate (Cys-FDH) = 1 × 103 M−1s−1) and the Cys-FDH variant’s slower kinetics was suggested to be due to a lower rate of the hydrogen atom transfer step (deuterium (formate) isotope effect on kcat/Km). Simultaneously, the pH-dependent alkylation-induced inactivation of the native SeCys-FDH and variant Cys-FDH (reaction with iodoacetamide in the presence of formate) was shown to follow the trend of the expected pKa values of each amino acid (native SeCys-FDH was inactivated more than 80% at pH > 6 (pKa (SeCys) ≈ 5.2), while variant Cys-FDH was inactivated more than 80% only at pH > 7 (pKa (Cys) ≈ 8.2). Together, these results were taken to suggest that selenol (versus thiol) plays an essential role in catalysis.
yme because of its thiol features.
As other variant enzymes are studied, it is becoming clear that it is not surprising that variants are less active than wild types. Most relevant to the present discussion was the recognition that several “wild-type variants” (native Cys-FDH) exist that are as catalytically efficient as the native SeCys-FDHs.
Selenocysteine incorporation is highly demanding (“expensive”) for the cell. It requires additional energy and dedicated machinery to uptake selenium and to synthesize and orchestrate different biomolecules that lead to the recognition of target UGA-codon by specific tRNA molecules (and not as the “opal” stop-codon), culminating in selenocysteine being incorporated in the target protein [102][103][104][105][106][102,103,104,105,106]. Therefore, it is generally accepted that the presence of selenocysteine should constitute an intrinsic advantage for the cell [14][107][108][109][14,107,108,109].
Regardless of the biological pressure behind the evolution of native SeCys-FDHs and native Cys-FDHs, it should be kept in mind that selenium is not a sulfur. Thus, it is reasonable that the presence of one or the other alters the reaction energy pattern, in spite of both enzyme types operating through the same general hydride transfer mechanism (same chemical transformations). Therefore, in order to be catalytically efficient, each enzyme type should have evolved a strategy to compensate for those Se/S physicochemical differences. Hence, more interesting and relevant than studying why some FDHs have selenium is to understand the strategies that allow both SeCys-FDH and Cys-FDH to be catalytically efficient.
4. Hydrogenases
Hases are crucial role as an alternative energy source as they have potential applications in green hydrogen production [110][111][112,113]. Hydrogenases are a heterogeneous group of enzymes that differ in size, subunit composition, metal content, and cellular location (periplasmic, cytoplasmic, and cytoplasmic membrane-bound) and catalyze the reversible two electron oxidation of hydrogen (Equation (2)). H2
⇌ 2H+
+ 2e−
4.1. Enzymatic Machineries
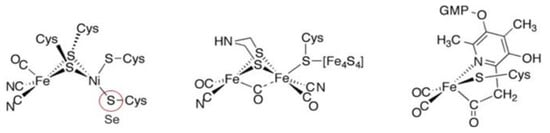
The metal-containing hydrogenases are subdivided into three classes: [Fe]-, [FeFe]-, and [NiFe]-hydrogenases (Figure 4) [110][111][112][113][114][115][112,113,114,115,116,117]. [Fe]-hydrogenases only contain one Fe ion in their active site and are designated as “Fe-only” hydrogenases. [FeFe]-hydrogenases contain an unusual iron-sulfur cluster termed the H-cluster that consists of an [Fe4S4] subcluster bridged via a cysteine (Cys) thiolate to the binuclear iron subcluster, also coordinated by inorganic ligands: two S atoms and one CO or CN ligand. [NiFe]-hydrogenases are heterodimeric proteins constituted by a small and a large subunit (Figure 5). The small subunit accommodates three iron-sulfur clusters (two [4Fe-4S] clusters and one [3Fe-4S] cluster) involved in the electron transport to/from the active site ([NiFe] cluster); the large subunit contains the catalytic site: the nickel-iron center. In some [NiFe]-hydrogenases, one of the Ni-bound cysteines is replaced by a selenocysteine, and [NiFe]- and [NiFeSe]-hydrogenases represent a single superfamily, and the Ni-Fe core contains unusual ligands: carbon monoxide (CO) and cyanide (CN−). Active site structure of [NiFe]-, [FeFe]-, and [Fe]-hydrogenases [112].
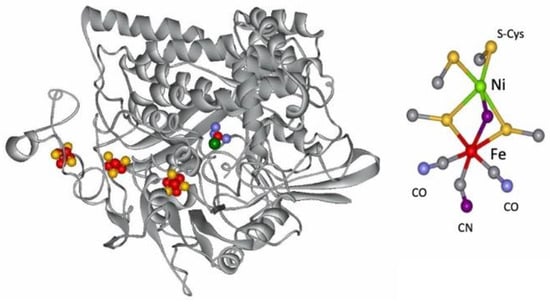
Figure 5.
Structure of the
D. gigas
hydrogenase enzyme and of its active site.
The [NiFe-Se] hydrogenases are found in some species of Desulfovibrio sp. The genes encoding the large and small subunits of the periplasmic hydrogenase from Desulfovibrio (D.) baculatus (DSM 1743) exhibit homology (40%) to the [NiFe] hydrogenases. The gene for the large subunit contains a codon (TGA) for selenocysteine in a position homologous to a codon (TGC) for cysteine in the [NiFe] hydrogenase. Spectroscopic studies support that selenium is a ligand to the nickel site (see below) [116][117][118][119][120][121][118,119,120,121,122,123].
As isolated, the active [NiFe] cluster contains a Ni(III) and a low-spin Fe(II) (diamagnetic) that remain unchanged during the enzyme mechanism. Different oxidized inactive states are attained by the enzyme. In general, the isolated states are mixtures of “unready” Ni-A and “ready” Ni-B states (Figure 6). These states show delocalized electron density between nickel and iron, attributed to a third bridging oxygenated ligand. Both oxidized states are paramagnetic and characterized by different EPR g-values. The bridging ligand in the Ni-B state has been assigned to an OH− ligand and a water molecule is probably present in the Ni-A state [122][123][124][125][124,125,126,127]. After the reaction with the substrate (hydrogen), (Ni-C) develops with a bridging hydride (H−) ligand.
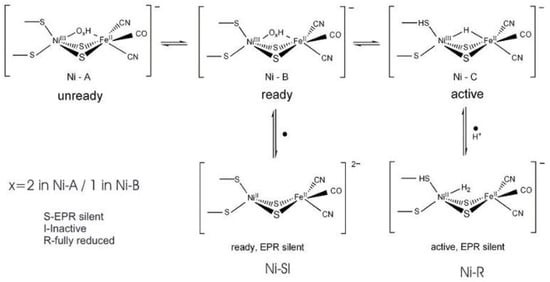
Figure 6. Redox and catalytic intermediates in [NiFe] hydrogenases. Adapted from [126].
4.2. Selenium and the Hydrogenase Reaction Mechanism
Isotopic substitutions are crucial for the identification of Ni and Se in hydrogenases. A
Redox and catalytic intermediates in [NiFe] hydrogenases. Adapted from [131].
4.2. Selenium and the Hydrogenase Reaction Mechanism
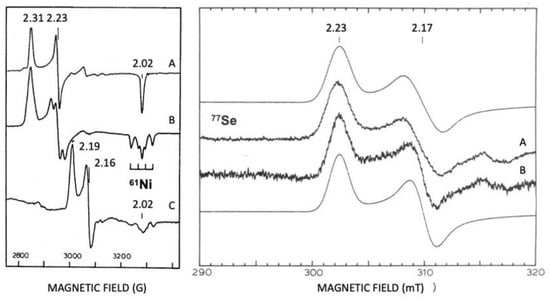
Isotopic substitutions are crucial for the identification of Ni and Se in hydrogenases. A Ni isotope was used for assigning EPR signals to Ni () [143]. Selenium contains six isotopes, and five of them are stable (atomic numbers 74, 76, 77, 78, and 80). The sixth isotope, with an atom abundance of 8.73%, is selenium-82 (Se), a beta emitter which is weakly radioactive. The Se isotope (7.5%) is a useful EPR marker, with an I = 1/2. Se and Se are useful markers for spectroscopic studies (EXAFS and EPR) [143,144,145,146].
61
Figure 7) [127]. Selenium contains six isotopes, and five of them are stable (atomic numbers 74, 76, 77, 78, and 80). The sixth isotope, with an atom abundance of 8.73%, is selenium-82 (
82
77
33
Figure 7.
Revealing EPR, Ni, and Se at the active site of hydrogenases. Isotopic substitutions with
61
Ni and
77
Se. Left panel
D. gigas
[Ni-Fe] Hase. (
A
) Ni-A
61
Ni un-enriched; (
B
) Ni-A
61
Ni enriched; (
C
) Ni-C
61
Ni enriched. Right panel
D. baculatus
[Ni-Fe-Se] Hase. (
A
) Ni-C
77
Se enriched and (
B
) Ni-C
Se un-enriched; mooth lines are simulations of spectra A and B. Adapted from refs [143,145].
Proton–deuterium exchange measurements are quite appropriate to probe the influence of the Se–cysteine ligand in the mechanism of hydrogen handling. An important clue was the observation that the H
2
/HD ratios were higher for [NiFeSe] hydrogenases than those observed for the [NiFe] ones, which is related to the activation of the hydrogen molecule (
Figure 8
).
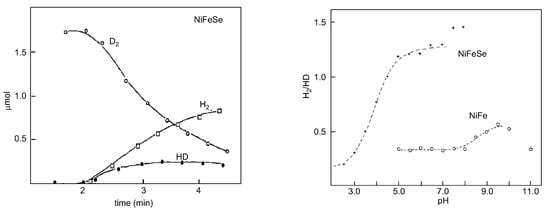
Figure 8.
D
2
/HD exchange activity of
D. salexigens
[NiFeSe] hydrogenase (
left panel
) and variation in the experimental ratios H
2
/HD as a function of pH (
right panel
) of
D. baculatus
(cytoplasmic) [NiFeSe] hydrogenase and
D. gigas [NiFe] (periplasmic). Left panel adapted from [131]; right panel adapted from [132].
4.3. Overview
Hydrogenases are a clear case study of the influence of Selenium (as a Se-Cys) on the modulating or fine-tuning of enzyme catalytic properties through an acid-base equilibrium at the proton acceptor site or at the hydride site and should be explored for protein design and molecular modelling [115].
The [NiFeSe] hydrogenases clearly emerge as a subgroup of [NiFe] and there is a structural homology between [NiFe] and [NiFeSe]. However, [NiFeSe] is distinct in terms of its catalytic and active-site composition. Electrochemical studies help to reveal the interplay between the catalytic intermediates [133]. These enzymes display very interesting catalytic properties for biological hydrogen production and bio-electrochemical applications: high H
[NiFe] (periplasmic). Left panel adapted from [140]; right panel adapted from [141].
4.3. Overview
Hydrogenases are a clear case study of the influence of Selenium (as a Se-Cys) on the modulating or fine-tuning of enzyme catalytic properties through an acid-base equilibrium at the proton acceptor site or at the hydride site and should be explored for protein design and molecular modelling [117].
The [NiFeSe] hydrogenases clearly emerge as a subgroup of [NiFe] and there is a structural homology between [NiFe] and [NiFeSe]. However, [NiFeSe] is distinct in terms of its catalytic and active-site composition. Electrochemical studies help to reveal the interplay between the catalytic intermediates [152]. These enzymes display very interesting catalytic properties for biological hydrogen production and bio-electrochemical applications: high H
2
production activity, low H
2
inhibition, and O
tolerance [153].
The direct role of selenocysteine in [NiFeSe] hydrogenase maturation and catalysis has also been discussed. An expression system for the production of recombinant [NiFeSe] hydrogenase from Desulfovibrio vulgaris Hildenborough and study of a selenocysteine–to-cysteine variant (Sec489Cys) in which, for the first time, a [NiFeSe] hydrogenase was converted to a [NiFe] type, reveal the direct involvement of this residue in the maturation process. It was proposed that selenium plays a crucial role in protecting against oxidative damage and the high catalytic activities of [NiFeSe] hydrogenases [135][133].
5. Glutathione Peroxidases
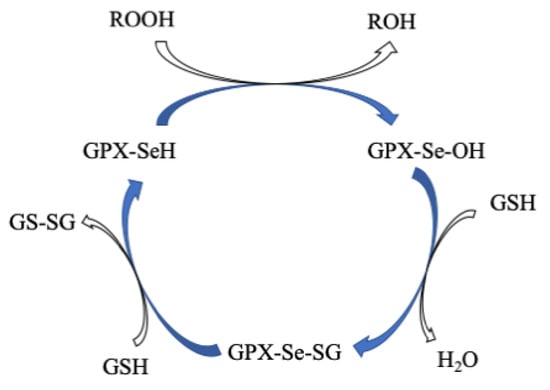
GPx is a multiple-isozyme family which protects the cellular organism from oxidative stress by the reductive transformation of hydroperoxide (H2O2) or organic hydroperoxide substrates (ROOH) to the product of H2O or alcohol, respectively, using cellular glutathione (GSH) as an electron source [136][137][154,155]. In 1952, Mills and Co-workers first noticed that GPX protected hemoglobin from oxidative degradation [138][156]. After that, in the 1960s, GPX activity was also observed in the lungs and kidneys [139][157]. In the 1970s, GPx was characterized and discovered selenocysteine amino acid, which played a vital role in enzymatic activity [140][141][142][158,159,160]. In the GPx family, only one GPx1 member was known until the 1980s. Then, this family grew to eight members [143][161]. All GPxs display two steps of redox reactions in their catalytic cycle (Figure 910) [144][145][171,172]. In the first step, the selenocysteine (Sec-SeH) is oxidized to selenic acid (Sec-SeOH), which is a key intermediate product in the catalytic cycle. Simultaneously, the toxic hydroperoxide is reduced to the corresponding alcohol. In the second step, the reduction of oxidized Sec-SeOH proceeds into two subsequent 1 e− reduction steps. The Sec-SeOH is converted into GPx-SeGS by interacting with one equivalent reduced GSH, followed by the reduction of GPx-SeGS into GPx-Se by a second equivalent GSH for the next catalytic cycle [138][140][146][147][148][156,158,173,174,175]. The intermediate Sec-SeOH is stabilized by Gln and Trp, which are in the catalytic tetrad site [149][170], and additional Asn in tetrad contributes to the catalytic reaction [150][167].Figure 910. Proposed catalytic cycle of GPxs. Modified from [138].
6. Thioredoxin Reductases
TrxR belongs to the pyridine nucleotide–disulfide oxidoreductases family, of which some members are glutathione reductase, mercuric ion reductase, and lipoamide dehydrogenase [151]. A homodimeric flavoenzyme, it contains one redox-active dithiol/disulfide motif, FAD prosthetic group, and an NADPH binding site in each monomeric subunit [152]. TrxRs are distributed in all living systems and are generally classified into two major classes: (a) low molecular weight (LMW~35 kDa) TrxRs that are present in both lower eukaryotes and prokaryotes, and (b) high molecular weight (HMW~55 kDa) TrxRs that are present in higher eukaryotes [151][153]. Both classes of TrxR utilize NADPH as an electron source to reduce the oxidized state of TrxR that plays a vital role in cell proliferation. Due to large differences in structures, both classes of TrxRs have different catalytic paths to execute the same biochemical reaction. The LMW TrxRs have two redox centers such as an
Proposed catalytic cycle of GPxs. Modified from [156].
6. Thioredoxin Reductases
TrxR belongs to the pyridine nucleotide–disulfide oxidoreductases family, of which some members are glutathione reductase, mercuric ion reductase, and lipoamide dehydrogenase [176]. A homodimeric flavoenzyme, it contains one redox-active dithiol/disulfide motif, FAD prosthetic group, and an NADPH binding site in each monomeric subunit [177]. TrxRs are distributed in all living systems and are generally classified into two major classes: (a) low molecular weight (LMW~35 kDa) TrxRs that are present in both lower eukaryotes and prokaryotes, and (b) high molecular weight (HMW~55 kDa) TrxRs that are present in higher eukaryotes [176,178]. Both classes of TrxR utilize NADPH as an electron source to reduce the oxidized state of TrxR that plays a vital role in cell proliferation. Due to large differences in structures, both classes of TrxRs have different catalytic paths to execute the same biochemical reaction. The LMW TrxRs have two redox centers such as an -terminal dithiol/disulfide pair and an FAD prosthetic group and sixteen additional amino acid residues with penultimate selenocysteine (Sec) in the catalytic site (-Cys-Secys-Gly sequence) at the end of the
-terminal [181,182,183,184].
-terminal dithiol/disulfide pair and an FAD prosthetic group [179,180], whereas HMW TrxRs contain three redox centers such as an
N
There are three types of Mammals’ TrxRs: (a) the cytosolic form, TrxR1 [160], (b) the mitochondrial form, TrxR2 [161][162], and (c) the testis-specific thioredoxin glutathione reductase (TGR) [163]. The overall protein fold of TrxR1 [164] is similar to other TrxR2 [165] and TGR [166]. Among them, TrxR1 is well-characterized. In 2001, the first three-dimensional (3D) structure of rat TrxR1 (Sec to Cys mutant) [164], followed by a large number of 3D structures (Sec-substituted mutants) of human TrxR1 [167] and mouse TrxR2 were published [165][168].
TrxR is an important biological redox mediator for the two-electron reduction of substrates. The catalytic cycle of mammalian TrxR involves three redox centers:
There are three types of Mammals’ TrxRs: (a) the cytosolic form, TrxR1 [185], (b) the mitochondrial form, TrxR2 [186,187], and (c) the testis-specific thioredoxin glutathione reductase (TGR) [188]. The overall protein fold of TrxR1 [189] is similar to other TrxR2 [190] and TGR [191]. Among them, TrxR1 is well-characterized. In 2001, the first three-dimensional (3D) structure of rat TrxR1 (Sec to Cys mutant) [189], followed by a large number of 3D structures (Sec-substituted mutants) of human TrxR1 [192] and mouse TrxR2 were published [190,193].
TrxR is an important biological redox mediator for the two-electron reduction of substrates. The catalytic cycle of mammalian TrxR involves three redox centers: -terminal dithiol (Cys–Cys), adjacent FAD/NADH, and -terminal selenolthiol pair (Cys–Sec) in the other subunit), which relay e from -terminal dithiol to the substrate, thioredoxin via FAD/NADH. The human TrxR1 substrate–thioredoxin (Trx) complex is identified and the 3D structure of that complex reveals that the -terminal arm binds with the substrate Trx through the disulphide bond (TrxR-Cys-S-S-Cys-Trx) [195].
N
59
64
C
498
497
−
N
C-terminal arm binds with the substrate Trx through the disulphide bond (TrxR-Cys-S-S-Cys-Trx) [169].
7. Iodothyronine Deiodinases
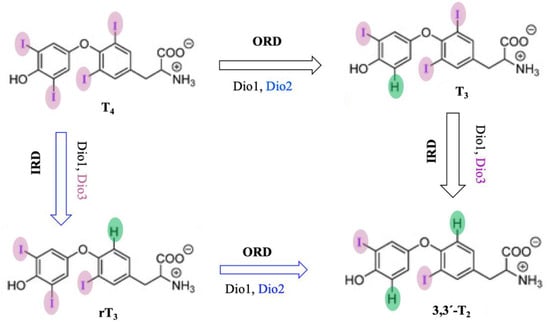
Dios are selenocysteine-dependent mammalian deiodinase enzymes that regulate thyroid hormones by deiodination of iodothyronine [170][171][172][173]. Dios have been classified into three isoforms, Dio1, Dio2, and Dio3, based on their sequence of amino acids and specificity of substrates. Dio1 enzymes non-selectively catalyze both inner- (phenolic group) and outer-ring (tyrosine group) deiodination of thyroid hormones, but Dio2 and Dio3 both selectively catalyze outer-ring and inner-ring deiodination (ORD and IRD) of thyroid hormones, respectively (
Dios are selenocysteine-dependent mammalian deiodinase enzymes that regulate thyroid hormones by deiodination of iodothyronine [199,200,201,202]. Dios have been classified into three isoforms, Dio1, Dio2, and Dio3, based on their sequence of amino acids and specificity of substrates. Dio1 enzymes non-selectively catalyze both inner- (phenolic group) and outer-ring (tyrosine group) deiodination of thyroid hormones, but Dio2 and Dio3 both selectively catalyze outer-ring and inner-ring deiodination (ORD and IRD) of thyroid hormones, respectively (3) [203,204,205,206,207,208,209].
Figure 103. Probable mechanism of deiodination by deiodinase with thyroid hormone substrates.
8. Selenoproteins and Human Health
8.1. Cancer
To date, several studies have attempted to analyze the role of GPxs, as well as changes in GPxs levels, in different types of tumors [181][182], but it remains controversial [183]. Indeed, GPx1 inhibits the oxidation of DNA mutations and, therefore, it may inhibit tumorigenesis [184], and overexpressed GPx1 reduces tumor growth, suggesting its protective effect in tumorigenesis [185]. However, reduced expression of GPx1 is detected in thyroid cancer [186], gastric cancer [187], and colorectal cancer [188], whereas GPx1 is highly expressed in kidney cancer [189] and pancreatic cancer [190]. Similar to GPx1, unusual expression of GPx2 is also observed in different tumors; for example, GPx2 is overexpressed in colorectal cancer [191], whereas a lower expression of GPx2 is detected in prostate intraepithelial neoplasia [192][193].
To date, several studies have attempted to analyze the role of GPxs, as well as changes in GPxs levels, in different types of tumors [216,220], but it remains controversial [221]. Indeed, GPx1 inhibits the oxidation of DNA mutations and, therefore, it may inhibit tumorigenesis [222], and overexpressed GPx1 reduces tumor growth, suggesting its protective effect in tumorigenesis [223]. However, reduced expression of GPx1 is detected in thyroid cancer [224], gastric cancer [225], and colorectal cancer [226], whereas GPx1 is highly expressed in kidney cancer [227] and pancreatic cancer [228]. Similar to GPx1, unusual expression of GPx2 is also observed in different tumors; for example, GPx2 is overexpressed in colorectal cancer [229], whereas a lower expression of GPx2 is detected in prostate intraepithelial neoplasia [230,231].
Polymorphism of human GPxs gene is a common phenomenon and it is associated with various diseases, especially tumors [194]. The GPx1 gene has various genetic polymorphisms and its most common polymorphism is the substitution of cytosine (C) to thymine (T) in DNA, resulting in the alteration of amino acid from proline (Pro) to leucine (Leu); thereby, the activity of GPx1 reduces by 5% [195]. Pro198Leu GPx1 polymorphism is associated with various types of cancer, mainly breast [196], prostate [197], lung [198], bladder [199], leukemia [200], and colon cancers [201]. However, the connection between GPx1 polymorphism and cancer vulnerability is controversial and inconclusive.
Polymorphism of human GPxs gene is a common phenomenon and it is associated with various diseases, especially tumors [248]. The GPx1 gene has various genetic polymorphisms and its most common polymorphism is the substitution of cytosine (C) to thymine (T) in DNA, resulting in the alteration of amino acid from proline (Pro) to leucine (Leu); thereby, the activity of GPx1 reduces by 5% [249]. Pro198Leu GPx1 polymorphism is associated with various types of cancer, mainly breast [250], prostate [251], lung [252], bladder [253], leukemia [254], and colon cancers [255]. However, the connection between GPx1 polymorphism and cancer vulnerability is controversial and inconclusive.
8.2. Diabetes
8.2. Diabetes
Diabetes mellitus (DM), a common human health problem around the globe, is a metabolic disorder and it is characterized by high levels of blood sugar (hyperglycemia), causing dysfunction in insulin secretion and/or sensitivity [276,277,278,279,280]. Insulin is a hormone synthesized in the β-cell of the pancreas and its action is also regulated by the pancreatic β-cell [280]. The most common type diabetes is Type 2 diabetes mellitus (T2DM) which is characterized by insulin resistance, caused by impairment of the pancreatic β-cell [280]. However, oxidative stress is believed to be the main cause of the onset and development of T2DM [281,282]. So, generation of ROS is a crucial factor in β-cell function [281].8.3. Viral Infections
Diabetes mellitus (DM), a common human health problem around the globe, is a metabolic disorder and it is characterized by high levels of blood sugar (hyperglycemia), causing dysfunction in insulin secretion and/or sensitivity [202][203][204][205][206]. Insulin is a hormone synthesized in the β-cell of the pancreas and its action is also regulated by the pancreatic β-cell [206]. The most common type diabetes is Type 2 diabetes mellitus (T2DM) which is characterized by insulin resistance, caused by impairment of the pancreatic β-cell [206]. However, oxidative stress is believed to be the main cause of the onset and development of T2DM [207][208]. So, generation of ROS is a crucial factor in β-cell function [207].
8.3. Viral Infections
Viral infections occur when the human body is invaded by viruses, such as human immunodeficiency virus (HIV) and severe acute respiratory syndrome—coronavirus 2 (SARSCoV2), that lead to many diseases. Viral infection often alters the intracellular redox homeostasis in the host cell by increasing ROS production, which enhances the viral replication [209][210][211][212][291,292,293,294]. Several selenoproteins, like glutathione peroxidases (GPxs) and thioredoxin reductase (TrxR), are important host antioxidants that may play an important role against viral infections by consuming ROS.