Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 1 by Preethi Chandrasekaran.
Non-alcoholic fatty liver disease (NAFLD) is a common and prevalent disorder affecting 25 percent of the adults in the United States and 32 percent of adults globally. It is one of the common causes of chronic liver disease characterized by steatosis, which can lead to inflammation, fibrosis, and cirrhosis. NAFLD is strongly associated with obesity and insulin resistance. Multiple genetic variants have been consistently found to be associated with NAFLD; one of them is found in the TMC4-MBOAT7 loci. One variant (rs641738 C>T) within MBOAT7 encoding lysophosphatidyl inositol acyltransferase increases the risk for NAFLD development and triggers hepatic inflammation by regulating arachidonic acid levels.
- lipid metabolism
- hepatic steatosis
- fibrosis
- therapeutic targets
- insulin resistance
- pathogenesis
- MBOAT7
1. Introduction
MBOA7 is a lysophophosphatidylinositol (LPI) acyltransferase preferentially transferring polyunsaturated fatty acids (PUFAs) to LPI. Emerging genome-wide association studies reported that a genetic variant within the MBOAT7 gene is closely related to non-alcoholic steatohepatitis (NASH), attributed to an increase in triglyceride synthesis through canonical and non-canonical pathways of de novo lipogenesis [9][1].
2. Mechanistic Link between MBOAT7 and Non-AFLDlcoholic Fatty Liver Disease and Related In Vivo/In Vitro Studies
It is crucial to further understand the molecular mechanisms underlying the progression of liver disease from simple steatosis to more advanced fibrotic disease. MBOAT7 association with NAFLD has emerged as a new lipid metabolic pathway as growing evidence suggests the pivotal role of MBOAT7 as a driver of NAFLD development and progression. Few studies have provided vital clues into its broader role and mechanistic insights.
MBOAT7 preferentially esterifies lysophosphatidylinositol (LPI) lipids to arachidonoyl-CoA to form major phosphoinositol (PI) species (38:4) in the inner leaflet of cell membranes. Phospholipases, prominently phospholipase A2 (PLA2), cleave fatty acid from the sn2 position and MBOAT7 selectively re-esterifies new PUFAs in that position, completing the remodeling cycle [31][2]. MBOAT7 loss of function could alter cellular signal transduction, protein–lipid interactions, vesicular transport, and membrane fusion events, given the fact that MBOAT7 generates the most abundant PI species (38:4) and key cellular phosphatidylinositol phosphates (PI 18:0/20:4) and PI ([18:0/20:4]-4,5P2). Another potential way by which MBOAT7 loss of function could promote NASH is by abnormal accumulation of LPI substrates in liver, as evidenced by multiple mice studies. These LPI substrates serve as relevant lipid signals promoting pro-inflammatory and pro-fibrotic effects [31][2].
The liver-specific knockout of MBOAT7 induces hepatic fat accumulation by increasing de novo lipogenesis driven by SREBP1, which is a key lipogenic transcription factor involved in fatty acid biosynthesis [22][3]. The non-canonical pathway, on the other hand, suggests that MBOAT7 depletion causes a simultaneous increase in PI synthesis and PI degradation mediated by a protein with PLC activity resulting in diacylglycerol, a substrate of triglyceride synthesis [32][4].
Lee and coworkers unraveled the first evidence of MBOAT7 enzymatic activity in Caenorhabditis elegans by RNA-interference-based genetic screening [33][5]. In C. elegans, eicosapentaenoic acid is the predominant PUFA, which is decreased by MBOAT7 deletion. It also exhibited reduced PI species and PI3P-related events [33][5]. Similarly, it has been shown that obese people have low levels of MBOAT7 in their livers and genetically modified obese mice with low MBOAT7 levels developed more severe NAFLD [8][6]. Strikingly, excess fat accumulation was noticed in human liver cells with low levels of MBOAT7. New approaches to therapeutic strategies in treating NAFLD in patients with MBOAT7 mutations can be developed with MBOAT7 being a critical mediator of NAFLD.
Several studies revealed that the hepatic MBOAT7 expression levels were suppressed in high-fat-diet mice and obese leptin-deficient mice and that MBOAT7 levels in adipose tissue were negatively correlated with insulin sensitivity and impaired glucose tolerance (Figure 1) [21,34][7][8].
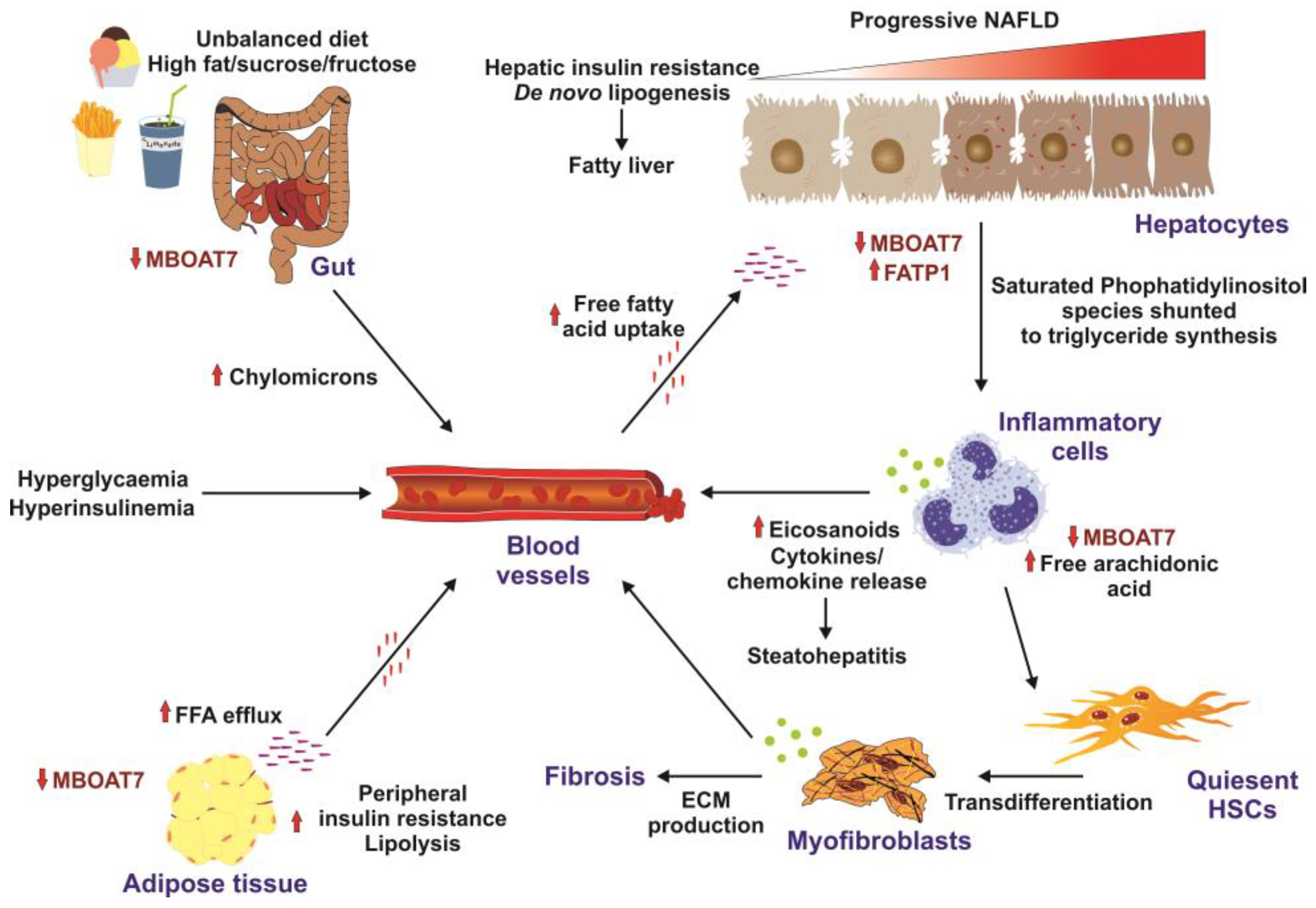
Figure 1. Factors promoting insulin resistance. High-caloric diets enriched in fat, sucrose, and fructose are risk factors for insulin resistance development. In the gut, the unbalanced diet results in downregulation of MBOAT7 and formation of chylomicrons. Elevated concentrations of systemic free fatty acids are taken up by hepatocytes, resulting in increased de novo lipogenesis and fatty liver. This provokes the infiltration of the liver with inflammatory cells, which results in elevated concentrations of cytokines and chemokines, provoking steatohepatitis and transdifferentiation of quiescent hepatic stellate cells (HSCs) to extracellular-matrix-producing (ECM) myofibroblasts leading to hepatic fibrosis. Similarly, to the liver and the gut, MBOAT7 is downregulated in the adipose, triggering peripheral insulin resistance. Solid arrows mark stimulatory effects. Small red lines indicate free fatty acids in the circulation, while small purple lines indicate tissue-bound free fatty acids. All these factors promote formation of hyperglycemia, hyperinsulinemia, and insulin resistance. This figure was redrawn in modified form from [34][8].
On a similar context, liver-specific deletion of MBOAT7 increased liver fat content in chow-diet-fed mice under fasting-re-feeding conditions. This hepatic lipid accumulation is shown to be caused due to an increase in de novo lipogenesis driven by SREBP1, supported by normalization of hepatic fat content by liver-specific deletion of MBOAT7 and SREBP cleavage-activating protein Scap [22][3].
In addition to the in vivo studies, Tanaka et al. investigated the impact of MBOAT7 deletion on PI content and fat accumulation in cultured hepatocytes. This restudy earch suggested that depletion of MBOAT7 in hepatocytes resulted in hepatic fat accumulation through increased triglyceride synthesis [32][4]. Here, it was proposed that hepatic lipid accumulation is due to a novel non-canonical pathway supplying substrates from PI to triglycerides through a futile cycle [32][4].
A wealth of recent data show that MBOAT7 overexpression in mice had beneficial effects on NASH pathology by significantly decreasing hepatic triglyceride levels and normalizing liver injury markers such as alanine aminotransferase and aspartate aminotransferase [35][9]. Longo et al. showed that MBOAT7 rs641738 and TM6SF2 E167k alters lipid droplet accumulation, mitochondrial morphology, and metabolic reprogramming towards HCC in vitro [36][10]. Strikingly, Krawczyk et al. confirmed the association of MBOAT7 with a more severe stage of fibrosis increasing plasma triglycerides, cholesterol, LDL, and glucose levels [37][11].
An interesting study by Raja et al. shed light on MBOAT7 rs641738 associations with NAFLD based on ethnicity [38][12]. It was shown that MBOAT7 is a strong contributor to the progression of NAFLD in the Caucasian and Chinese population, while it is not significant in Afro-American and Hispanic populations [39][13]. Yet another unique finding by Massey et al. showed diet-induced metabolic disturbances, hyperinsulinemia, and systemic insulin resistance in mice with adipocyte-specific disruption of the MBOAT7 gene [21][7].
Furthermore, liver-specific knockdown of MBOAT7 by antisense oligonucleotides revealed large alterations in liver lipid storage characterized by the accumulation of triglycerides, free cholesterol, and cholesterol esters in high-fat-diet-fed mice [8][6]. In addition, MBOAT7 knockdown was associated with liver injury as indicated by elevated aspartate aminotransferase and alanine aminotransferase in these animals. Aligned with these alterations, knockdown of MBOAT7 in high-fat-diet-fed mice resulted in alterations in LPI and PI lipids in a tissue-specific manner such as selective reduction in 38:3 and 38:4 species of circulating PI lipids and significant accumulation of 16:0 and 18:1 LPI species in the liver, inducing an imbalance of local lipid mediators that originate from PI metabolism.
A recent meta-analysis of 42 studies including more than 1 million participants showed that the MBOAT7 variation is firmly associated with the severity of NAFLD in European adults [40][14]. Another interesting study suggests that MBOAT7 is a negative regulator of TLR signaling and highlights that MBOAT7 modulation can be beneficial for suppressing inflammation associated with the dysregulation of Toll-like receptor signaling such as metabolic-associated fatty liver diseases (MAFLD) [24][15].
3. Challenges and Recent Progress in Treating Complex Non-AFLlcoholic Fatty Liver Disease
NAFLD represents a ‘silent epidemic’ as it is often asymptomatic in nature with increased prevalence among adults with obesity, type 2 diabetes, insulin resistance, and metabolic syndrome [41][16]. It represents a growing public health challenge owing to lack of approved therapies at present [42][17]. Individuals with NAFLD present at least one feature of metabolic syndrome, making it an even more challenging multi-systemic disease. In addition, the complexity is increased by the fact that many patients remain undiagnosed in early phases of the disease [43][18]. Variability in NAFLD-related risk factors, substantial mortality, and morbidity are a few other challenges in the management of this disease [43][18]. Therefore, it is important to adopt a holistic approach in managing this diversified condition.
At present, NAFLD is treated with lifestyle modifications such as weight loss and dietary changes as there is no approved therapy yet for this multi-factorial disease [42][17]. Multiple drug strategies are being developed and tested to treat advanced NAFLD and target inflammatory, fibrotic, and metabolic pathways. More recently, a structure and model were reported for the catalytic mechanism of human MBOAT7, which reveals a twisted tunnel from the cytosol and luminal side providing access for arachidonoyl-CoA and lyso-PI. This structure might be important in the identification of small molecule inhibitors for targeted drug therapy in treating NAFLD [12][19].
There is one other study, which elucidated that increased expression of MBOAT7 is co-related with detrimental outcomes in HCC, emphasizing the role of MBOAT7 inhibitors as useful therapeutic targets in treating HCC [44][20].
A profound study by Thangapandi et al. provided combined mice and human datasets and demonstrated that targeting PI signaling might be a potential therapeutic option for treating NAFLD and fibrosis. Their study unfolded a novel finding that MBOAT7 deficiency in mice and humans points to an inflammation-independent pathway of liver fibrosis mediated by lipid signaling, opening new avenues for potential targets for NAFLD [45][21]. It was also shown via lipidomics that in addition to PI and LPI, phosphoglycerol, lysophophatidylglycerol, and phosphatidic acids were increased in MBOAT7-deficient livers.
For patients with biopsy-proven NASH, vitamin E and pioglitazone supplements are recommended, although there are concerns regarding side effects [46][22]. As previously mentioned, intestinal microbiota play an important role in NAFLD pathogenesis. Thus, probiotics, antibiotics, and prebiotics might play a therapeutic role via modulating gut microbiota [47][23].
Genetic screening for polymorphisms to identify the individuals at high risk for NAFLD will help in deeper understanding of specific therapeutic strategies. Modulators of bile acid signaling medications to improve insulin sensitivity are being tested in patients with NASH as a possible therapeutic approach [48][24].
Currently, there are several innovative pipeline drugs that are tested in clinical trials for the treatment of NAFLD/NASH (Figure 2).
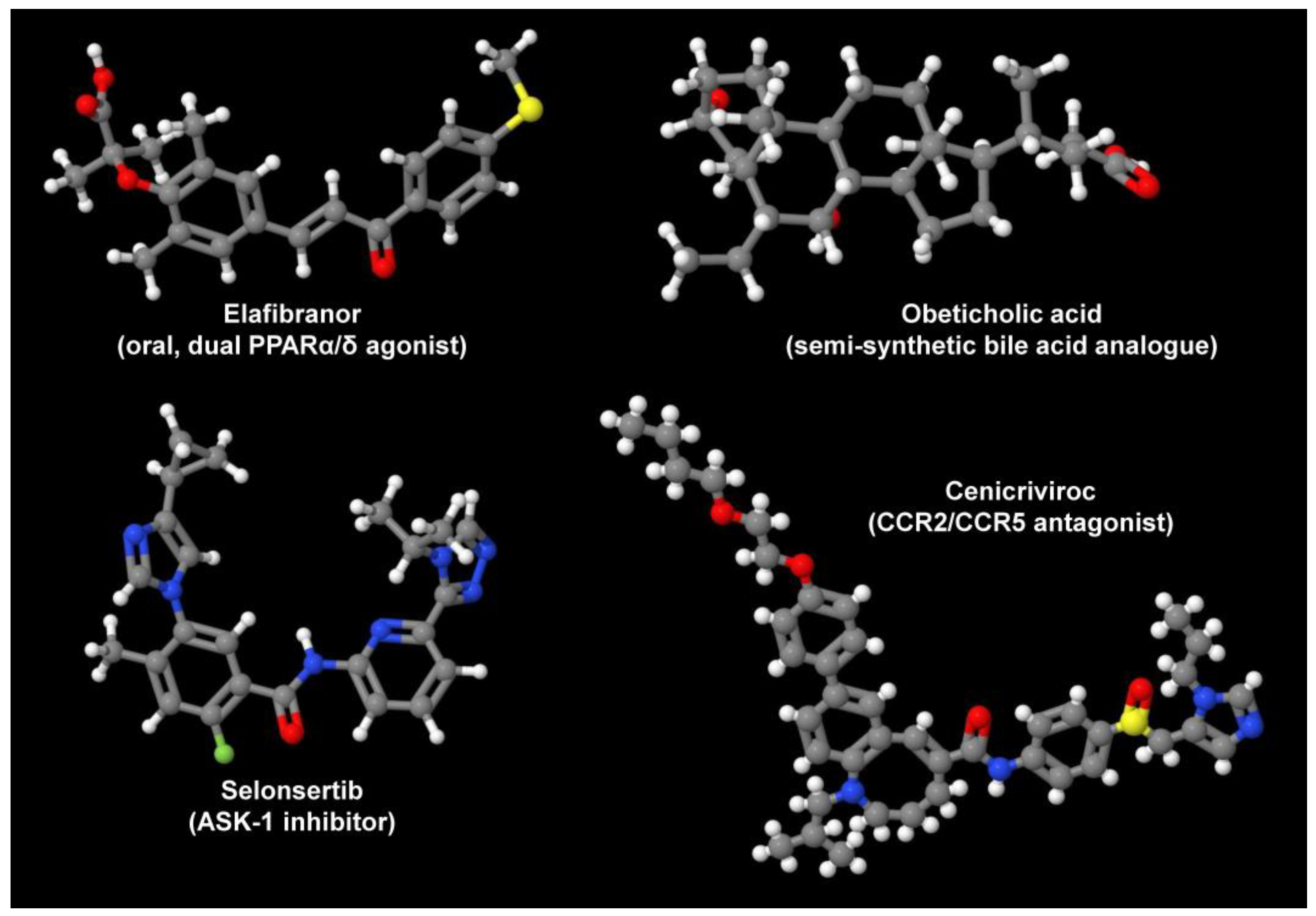
Figure 2. Drug candidates for treatment of NAFLD/NASH. Depicted are the oral, dual PPARα/δ agonist Elafibranor, the semi-synthetic bile acid analogue obeticholic acid, the ASK-1 inhibitor Selonsertib, and the CCR2/CCR5 antagonist Cenicriviroc. These are only some drugs that are tested in ongoing studies. In addition, there are several TLR4 antagonists and macrolide antibiotics that are tested in ongoing trials. Their variable mode of action demonstrates that there are many independent options to target the pathogenesis of NAFLD/NASH. All structures depicted were generated with the open-source molecule viewer Jmol, version 14.2.15_2015.07.09 [49][25] using information depicted in PubChem [50][26] for Elafibranor (PubChem CID: 9864881), obeticholic acid (PubChem CID: 447715), Selonsertib (PubChem CID: 71245288), and Cenicriviroc (PubChem CID: 11285792), respectively.
Elafibranor, which acts as a dual-peroxisome proliferation-activated receptor (PPAR) α/δ agonist, improves glucose metabolism and insulin sensitivity and decreases inflammation. In a phase 2b randomized control study (NCT0164849, GOLDEN-505), the effects of Elafibranor (120.80 mg/day) were studied for 52 weeks. NASH was resolved in 19% of patients compared to 9% in the placebo group. In addition, a phase 3 RCT (NCT02704403, RESOLVE-IT) is being evaluated on the effects of Elafibranor (120 mg/day for 72 weeks). Additional novel PPAR agonists such as Saroglitazar and Lanifibranor are also being tested [51][27].
Obeticholic acid, a semi-synthetic bile activator of Farnesoid X receptors, regulates lipid/glucose homeostasis, promotes insulin sensitivity, and modulates liver fibrosis. In a phase 2b RCT (NCT01264598, FLINT), obeticholic acid (25 mg/day) was tested for 72 weeks in patients with a NAFLD score greater than 4. Remarkably, 45% of the patients showed histological improvement compared with 21% in the placebo group [52][28]. This trial progressed to a phase 3 RCT (NCT02548351 REGENERATE), which is currently recruiting patients with biopsy-proven NASH to assess obeticholic acid’s effects and to evaluate the long-term effects over a 7-year period [53][29].
The involvement of inflammatory cells and cascade of inflammatory events is well known in hepatocyte injury. Cenicriviroc (CVC) functions as a dual antagonist of CCR2 and CCR5 and demonstrated decreased fibrosis in preclinical models. A phase 2 RCT (NCT02217475, CENTAUR) with CVC 150 mg/day was evaluated in 289 patients with NASH, fibrosis, and diabetes mellitus. Although there was no significant improvement in NASH after 12 months, liver fibrosis improved in 20% patients as compared to 10% in the placebo group. A phase 3 RCT (NCT03028740 AURORA) was initiated to evaluate the effects of 150 mg/day CVC with long-term follow-up over a 5-year period [54][30].
TNF-α signaling plays an important role in hepatocyte injury and apoptosis, which in turn activates the apoptosis signal-regulating kinase 1 (ASK1) leading to hepatic inflammation, fibrosis, and hepatocyte apoptosis. Selonsertib, a selective inhibitor of ASK1, was tested for its effects on patients with NASH and fibrosis in a phase 2 RCT (NCT02466516). The patients showed fibrosis improvement after 24 weeks of treatment. Two phase 3 trials (NCT03053050 STELLAR3, NCT03053063 STELLAR 4) are being conducted currently to evaluate the effects of Selonsertib at week 48 and further monitor at 240 weeks [55][31].
In principle, there are also ways to directly target MBOAT7 gene expression or activity. Since the suppression of MBOAT7 was shown to drive hepatic fat accumulation and NAFLD development [8[6][32],56], the overexpression of MBOAT7 or approaches leading to increased endogenous expression of MBOAT7 should have beneficial effects on the outcome of NAFLD. Therapeutic gene therapy has been used in a plethora of diseases so far [57][33]. In particular, engineered hepatotropic adeno-associated viruses, retroviral vectors, or lentiviral delivery systems expressing MBOAT7 under transcriptional control of liver-specific promoters could be used to increase the overall concentration of MBOAT7 in the liver. Similarly, the transfer of nanoparticles, liposomes, polymers, virus-like particles, erythrocyte ghosts, and exosomes that are loaded with MBOAT7 expression constructs, or special in vivo or ex vivo gene transfer techniques might be scalable alternatives to increase MBOAT quantities [57][33].
Similarly, strategies that enhance the translation and stability of endogenous or exogenous mRNA that were already used in other disease scenarios could be applied [58][34]. Finally, in the long term, there will be gene replacement techniques available (e.g., CRISPR/Cas9) that will allow for the replacement of MBOAT7 mutations that are associated with increased accumulation of intracellular free fatty acids and hepatic steatosis. Nevertheless, although gene therapy is a promising therapeutic strategy that made remarkable advancements during the last decade, there are still many hurdles in the use of this promising therapy [59][35].
Nevertheless, a proof-of-concept study has recently shown that the overexpression of MBOAT7 in mice fed either a choline-deficient high-fat diet or a Gubra Amylin NASH diet and subsequent infected with an adeno-associated virus expressing MBOAT7 failed to improve in terms of NASH pathology [35][9]. However, in the mentioned study, the authors demonstrated that MBOAT7 overexpression slightly improved liver weights, triglycerides, and plasma alanine and aspartate transaminases [35][9]. It is possible that MBOAT7 needs additional factors to be therapeutically effective in NAFLD/NASH. This again highlights the complexity of the NAFLD/NASH pathogenesis that is driven by many genetic and epigenetic factors and pinpoints the fact that further studies are urgently needed to identify proper targeted therapies for NALFD/NASH.
References
- Tavaglione, F.; Kono, N.; Romeo, S. Understanding the underlying molecular pathways by which MBOAT7/Lpiat1 depletion induces hepatic steatosis. J. Lipid Res. 2021, 62, 100047.
- Varadharajan, V.; Massey, W.J.; Brown, J.M. Membrane-bound O-acyltransferase 7 (MBOAT7)-driven phosphatidylinositol remodeling in advanced liver disease. J. Lipid Res. 2022, 63, 100234.
- Xia, M.; Chandrasekaran, P.; Rong, S.; Fu, X.; Mitsche, M.A. Hepatic deletion of MBOAT7 (LPIAT1) causes activation of SREBP-1c and fatty liver. J. Lipid Res. 2021, 62, 100031.
- Yuki, T.; Yuta, S.; Andrea, C.; Takuya, K.; Yanli, M.; Tetsuya, K.; Naoto, K.; Toshimasa, Y.; Rosellina Margherita, M.; Guido, B.; et al. LPIAT1/MBOAT7 depletion increases triglyceride synthesis fueled by high phosphatidylinositol turnover. Gut 2021, 70, 180.
- Lee, H.-C.; Inoue, T.; Imae, R.; Kono, N.; Shirae, S.; Matsuda, S.; Gengyo-Ando, K.; Mitani, S.; Arai, H. Caenorhabditis elegans mboa-7, a member of the MBOAT family, is required for selective incorporation of polyunsaturated fatty acids into phosphatidylinositol. Mol. Biol. Cell 2007, 19, 1174–1184.
- Helsley, R.N.; Varadharajan, V.; Brown, A.L.; Gromovsky, A.D.; Schugar, R.C.; Ramachandiran, I.; Fung, K.; Kabbany, M.N.; Banerjee, R.; Neumann, C.K.; et al. Obesity-linked suppression of membrane-bound O-acyltransferase 7 (MBOAT7) drives non-alcoholic fatty liver disease. eLife 2019, 8, e49882.
- Massey, W.J.; Varadharajan, V.; Banerjee, R.; Brown, A.L.; Horak, A.J.; Hohe, R.C.; Jung, B.M.; Qiu, Y.; Chan, E.R.; Pan, C.; et al. MBOAT7-driven lysophosphatidylinositol acylation in adipocytes contributes to systemic glucose homeostasis. J. Lipid Res. 2023, 64, 100349.
- Meroni, M.; Longo, M.; Fracanzani, A.L.; Dongiovanni, P. MBOAT7 down-regulation by genetic and environmental factors predisposes to MAFLD. eBioMedicine 2020, 57, 102866.
- Sharpe, M.C.; Pyles, K.D.; Hallcox, T.; Kamm, D.R.; Piechowski, M.; Fisk, B.; Albert, C.J.; Carpenter, D.H.; Ulmasov, B.; Ford, D.A.; et al. Enhancing hepatic MBOAT7 expression in mice with nonalcoholic steatohepatitis. Gastro Hep Adv. 2023, 2, 558–572.
- Longo, M.; Meroni, M.; Paolini, E.; Erconi, V.; Carli, F.; Fortunato, F.; Ronchi, D.; Piciotti, R.; Sabatini, S.; Macchi, C.; et al. TM6SF2/PNPLA3/MBOAT7 Loss-of-function genetic variants impact on NAFLD development and progression both in patients and in in vitro models. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 759–788.
- Krawczyk, M.; Rau, M.; Schattenberg, J.M.; Bantel, H.; Pathil, A.; Demir, M.; Kluwe, J.; Boettler, T.; Lammert, F.; Geier, A. Combined effects of the PNPLA3 rs738409, TM6SF2 rs58542926, and MBOAT7 rs641738 variants on NAFLD severity: A multicenter biopsy-based study. J. Lipid Res. 2017, 58, 247–255.
- Raja, A.M.; Ciociola, E.; Ahmad, I.N.; Dar, F.S.; Naqvi, S.M.S.; Moaeen-Ud-Din, M.; Kaukab Raja, G.; Romeo, S.; Mancina, R.M. Genetic susceptibility to chronic liver disease in individuals from Pakistan. Int. J. Mol. Sci. 2020, 21, 3558.
- Sookoian, S.; Flichman, D.; Garaycoechea, M.E.; Gazzi, C.; Martino, J.S.; Castaño, G.O.; Pirola, C.J. Lack of evidence supporting a role of TMC4-rs641738 missense variant—MBOAT7- intergenic downstream variant—In the susceptibility to nonalcoholic fatty liver disease. Sci. Rep. 2018, 8, 5097.
- Teo, K.; Abeysekera, K.W.M.; Adams, L.; Aigner, E.; Anstee, Q.M.; Banales, J.M.; Banerjee, R.; Basu, P.; Berg, T.; Bhatnagar, P.; et al. rs641738C>T near MBOAT7 is associated with liver fat, ALT and fibrosis in NAFLD: A meta-analysis. J. Hepatol. 2021, 74, 20–30.
- Alharthi, J.; Bayoumi, A.; Thabet, K.; Pan, Z.; Gloss, B.S.; Latchoumanin, O.; Lundberg, M.; Twine, N.A.; McLeod, D.; Alenizi, S.; et al. A metabolic associated fatty liver disease risk variant in MBOAT7 regulates toll like receptor induced outcomes. Nat. Commun. 2022, 13, 7430.
- Sivell, C. Nonalcoholic fatty liver disease: A silent epidemic. Gastroenterol. Nurs. 2019, 42, 428–434.
- Meroni, M.; Longo, M.; Rustichelli, A.; Dongiovanni, P. Nutrition and genetics in NAFLD: The perfect binomium. Int. J. Mol. Sci. 2020, 21, 2986.
- Arab, J.P.; Díaz, L.A.; Dirchwolf, M.; Mark, H.E.; Lazarus, J.V.; Vaughan, E.; Méndez-Sánchez, N.; Oliveira, C.P.; Gadano, A.; Arrese, M. NAFLD: Challenges and opportunities to address the public health problem in Latin America. Ann. Hepatol. 2021, 24, 100359.
- Wang, K.; Lee, C.-W.; Sui, X.; Kim, S.; Wang, S.; Higgs, A.B.; Baublis, A.J.; Voth, G.A.; Liao, M.; Walther, T.C.; et al. The structure of phosphatidylinositol remodeling MBOAT7 reveals its catalytic mechanism and enables inhibitor identification. Nat. Commun. 2023, 14, 3533.
- Donati, B.; Dongiovanni, P.; Romeo, S.; Meroni, M.; McCain, M.; Miele, L.; Petta, S.; Maier, S.; Rosso, C.; De Luca, L.; et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 2017, 7, 4492.
- Thangapandi, V.R.; Knittelfelder, O.; Brosch, M.; Patsenker, E.; Vvedenskaya, O.; Buch, S.; Hinz, S.; Hendricks, A.; Nati, M.; Herrmann, A.; et al. Loss of hepatic MBOAT7 leads to liver fibrosis. Gut 2021, 70, 940–950.
- Bril, F.; Biernacki, D.M.; Kalavalapalli, S.; Lomonaco, R.; Subbarayan, S.K.; Lai, J.; Tio, F.; Suman, A.; Orsak, B.K.; Hecht, J.; et al. Role of vitamin E for nonalcoholic steatohepatitis in patients with type 2 diabetes: A randomized controlled trial. Diabetes Care 2019, 42, 1481–1488.
- Iacono, A.; Raso, G.M.; Canani, R.B.; Calignano, A.; Meli, R. Probiotics as an emerging therapeutic strategy to treat NAFLD: Focus on molecular and biochemical mechanisms. J. Nutr. Biochem. 2011, 22, 699–711.
- Fiorucci, S.; Biagioli, M.; Sepe, V.; Zampella, A.; Distrutti, E. Bile acid modulators for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin. Investig. Drugs 2020, 29, 623–632.
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D with Features for Chemicals, Crystals, Materials and Biomolecules. Available online: https://jmol.sourceforge.net/ (accessed on 9 November 2023).
- PubChem. National Library of Medicine. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 9 November 2023).
- Lange, N.F.; Graf, V.; Caussy, C.; Dufour, J.F. PPAR-targeted therapies in the treatment of non-alcoholic fatty liver disease in diabetic patients. Int. J. Mol. Sci. 2022, 23, 4305.
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965.
- Carr, R.M.; Reid, A.E. FXR agonists as therapeutic agents for non-alcoholic fatty liver disease. Curr. Atheroscler. Rep. 2015, 17, 500.
- Lefebvre, E.; Moyle, G.; Reshef, R.; Richman, L.P.; Thompson, M.; Hong, F.; Chou, H.L.; Hashiguchi, T.; Plato, C.; Poulin, D.; et al. Antifibrotic effects of the dual CCR2/CCR5 antagonist Cenicriviroc in animal models of liver and kidney fibrosis. PLoS ONE 2016, 11, e0158156.
- Harrison, S.A.; Wong, V.W.; Okanoue, T.; Bzowej, N.; Vuppalanchi, R.; Younes, Z.; Kohli, A.; Sarin, S.; Caldwell, S.H.; Alkhouri, N.; et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J. Hepatol. 2020, 73, 26–39.
- Meroni, M.; Dongiovanni, P.; Longo, M.; Carli, F.; Baselli, G.; Rametta, R.; Pelusi, S.; Badiali, S.; Maggioni, M.; Gaggini, M.; et al. MBOAT7 down-regulation by hyper-insulinemia induces fat accumulation in hepatocytes. eBioMedicine 2020, 52, 102658.
- Sayed, N.; Allawadhi, P.; Khurana, A.; Singh, V.; Navik, U.; Pasumarthi, S.K.; Khurana, I.; Banothu, A.K.; Weiskirchen, R.; Bharani, K.K. Gene therapy: Comprehensive overview and therapeutic applications. Life Sci. 2022, 294, 120375.
- Kairuz, D.; Singh, P.; Smith, T.; Arbuthnot, P.; Ely, A.; Bloom, K. Synthetic mRNA gene therapies and hepatotropic non-viral vectors for the treatment of chronic HBV infections. In Messenger RNA Therapeutics, RNA Technologies; Jurga, S., Barciszewski, J., Eds.; Springer: Cham, Switzerland, 2022; Volume 13.
- Khurana, A.; Sayed, N.; Singh, V.; Khurana, I.; Allawadhi, P.; Rawat, P.S.; Navik, U.; Pasumarthi, S.K.; Bharani, K.K.; Weiskirchen, R. A comprehensive overview of CRISPR/Cas 9 technology and application thereof in drug discovery. J. Cell. Biochem. 2022, 123, 1674–1698.
More