1. Childhood Obesity Definition and BMI Related CKD Progression
In adults, the body mass index (BMI) serves as the standard measure to identify obesity as a health risk factor. However, the criteria for classifying overweight and obese individuals differ for children. According to the Centers for Disease Control, overweight children are those with a BMI above the 85th percentile, while obese children have a BMI > 95th percentile
[1][17]. The American Heart Association defines severe obesity as having a BMI ≥ 120% of the 95th percentile or an absolute BMI ≥ 35 kg/m
2 [2][18]. The baseline BMI has emerged as a critical predictor of CKD progression
[3][19]. Research indicates that each unit increase in BMI heightens the risk of ESRD
[4][20]. An observational study by Russo et al. underlines a significant link between high BMI and the initiation of dialysis in CKD patients. It suggested that hemodynamic changes may explain this increased risk as high BMI triggers glomerular hypertrophy and subsequent hyperfiltration, potentially expediting renal function decline and necessitating earlier dialysis initiation
[5][21].
2. Impact of Obesity on Kidney Outcomes in Children
While the effects of obesity on renal function in adults have been extensively studied and well-documented, recent research is shedding light on similar trends in children. A pivotal 2017 systematic review and meta-analysis by Garofalo et al., involving over 600,000 participants from 39 cohort studies, conclusively linked obesity to an increased risk of low glomerular filtration rate (GFR) and albuminuria over a mean follow-up period of 6.8 years in adults
[6][22]. This landmark study established BMI as an independent predictor of de novo CKD in the general adult population
[6][22]. Further studies have reinforced these findings, emphasizing the association between abdominal obesity and the high risk of all-cause and CV mortality among ESRD patients
[7][23]. Additionally, another large study conducted over nearly two decades (1964 to 1985) involving 330,252 individuals in the state of California in the U.S.A. demonstrated that a high BMI is a robust and modifiable risk factor for ESRD, even after adjustments for baseline blood pressure (BP) levels and the presence of diabetes mellitus (DM)
[8][24].
In children with CKD, the evidence, while not as extensive as that in adults, suggests a similar trend. Children with obesity often become adults with obesity, thereby increasing the risk of various clinical outcomes, particularly metabolic diseases
[9][10][25,26]. Obesity plays a significant role in both the onset and the progression of CKD
[11][12][27,28] and is strongly correlated with the two most common causes of ESRD: DM and hypertension
[13][29]. Metabolic syndrome, a consequence of obesity, also stands out as an independent risk factor for ESRD
[14][30]. Notably, bariatric surgery has consistently demonstrated its ability to reduce the prevalence of metabolic syndrome
[15][31]. In a study with a median follow-up of 13 years, children with DM had a 154% increased risk of early-onset kidney disease compared with their counterparts without DM
[16][32]. In addition, diabetic children have an increased risk of other specific kidney diseases, including tubulo-interstitial and glomerular diseases, urolithiasis, and renal failure. The risk of developing these conditions tends to be higher in children with type 2 DM than in those with type 1-DM
[16][32]. A consistent association has been demonstrated between increased BMI in adolescents and the development of ESRD from diabetic and nondiabetic causes. In a large study of 1.2 million adolescents with a median follow-up of about 25 years, obese adolescents showed a 3.4-fold increased risk of developing ESRD from nondiabetic nephropathy and a 19-fold increased risk of developing ESRD from diabetic causes
[17][13].
Differently from adults, obesity-related kidney damage is believed to occur early in childhood, long before the onset of hypertension, DM, and other comorbidities commonly associated with kidney disease. Thus, obese children have been seen to have larger kidneys and increased renal flow compared with normal-weight children, supporting the idea that obesity may trigger renal changes very early in life
[18][16].
Recent studies underscore that obesity is an independent risk factor for CKD in children
[19][20][33,34], contributing to an increased risk of death among obese children with ESRD
[21][22][35,36]. Moreover, even otherwise healthy overweight or obese children exhibit a markedly increased risk of ESRD later in life
[17][23][13,37]. The long-term impact of overweight and obesity on CKD risk is substantial, as demonstrated by a large prospective study with 4463 participants, revealing that early-onset overweight between the ages of 26 and 36 was strongly associated with reduced renal function at age 60–64 years
[24][38].
In one Canadian tertiary pediatric clinic, nephrology patients referred between 1985 and 2006 displayed significantly higher BMI z-scores, with obese patients having a higher risk of developing CKD later in life, surpassing predictions based solely on their primary kidney disease
[25][39].
Surprisingly, despite the usual presence of poor nutritional status in patients undergoing renal replacement therapy, a significant proportion of the European pediatric population undergoing renal replacement therapy is overweight or obese, rather than underweight
[26][14]. This finding carries significant health implications, as obese children with ESRD and a high BMI have an increased risk of death
[27][40]. Therefore, nutritional management in children and adolescents in renal replacement therapy should be focused on the prevention and treatment of overweight and obesity over malnutrition. Considering these aforementioned factors, pediatric nephrologists are increasingly recognizing obesity as a serious health concern
[22][36].
3. Hereditary Factors of Obesity
Obesity is a complex disease influenced by both genetic (multiple allelic variants) and environmental factors (e.g., more calorie-dense food and increasingly sedentary lifestyles)
[28][29][41,42]. The interactions between genes and an increasingly obesogenic environment play a crucial role in the physiology and pathophysiology of obesity. Body adiposity has a hereditary component, but the identification of specific genes responsible for common forms of obesity has proven to be challenging. However, genomic analyses have shed light on allelic variants associated with obesity. Environmental factors, such as lifestyle changes, dietary shifts (including increased intake of sugary foods and drinks), and reduced physical activity
[30][43], seem to modify genetic associations with BMI. Notably, severe early-onset obesity is more likely to have a predominantly genetic origin.
In cases of monogenic forms of obesity, where genetic mutations significantly contribute to obesity, targeted therapies may be considered as part of the treatment. For example, the variants of the fat mass and obesity-associated gene (
FTO), located on chromosome 16, exemplify the interplay between genetics and environment
[31][32][44,45]. Although the exact mechanisms linking
FTO genes to obesity remain unclear, studies indicate that they primarily affect exergy expenditure. Additionally, certain genetic variants can modify adipocyte function, influencing energy utilization and mitochondrial thermogenesis
[33][46]. This observation has important implications for the discovery of new anti-obesity drugs. The effects of single nucleotide polymorphisms on adiposity are influenced by physical activity and macronutrient composition in the diet
[34][47]. Other hereditary factors can independently increase the risk of conditions such as DM through various effects on different tissues
[35][48].
Several specific hereditary factors can contribute to obesity, especially in children (Figure 1):
Figure 1. Graphical representation of some of the most important and known hereditary factors of obesity.
- I.
-
leptin: Produced by adipose cells in the placenta and, to a lesser extent, in the intestine. The
Ob (Obese) or
Lep (Leptin) gene codes for leptin, which signals to the brain, regarding the levels of stored fat in the body. Leptin-deficient mice (
Ob mice) show hyperphagia, insulin resistance, hyperinsulinemia, and infertility. By increasing adiposity, resistance to the action of leptin occurs. Although obesity due to leptin deficiency has been studied, most people with obesity do not have any abnormalities in the leptin gene. This suggests that obesity may be caused by either leptin deficiency or genetic defects in the leptin receptor itself
[36][37][49,50].
-
- II.
-
Prohormone convertase 1/3 (PC1/3): a congenital deficiency of the
PCSK1 gene, responsible for proprotein convertase 1/3, can lead to a severe multihormonal disorder characterized by early-onset obesity
[38][51].
-
- III.
-
Congenital deficiency of the melanocortin-4 receptor (MC
4R): a congenital deficiency of MC
4R is associated with early-onset obesity and above-average height
[39][40][41][52,53,54].
-
- IV.
-
Proopiomelanocortin (POMC) or melanocyte-stimulating hormone (MSH): It transmits the appetite-suppressing effect of leptin through MC
4R. Mutations in
POMC gene can also lead to early-onset obesity due to severe hyperphagia. ACTH (AdrenoCorticoTropic Hormone) is produced by POMC in the hypothalamus, as is alpha-MSH, a key factor in reducing food intake
[42][43][55,56].
-
- V.
-
Guanine nucleotide-binding protein G, alpha stimulant
(GNAS) gene mutations: these are associated with Albright’s hereditary osteodystrophy (i.e., type 1 pseudohypoparathyroidism), and characterized by early-onset obesity, along with other features such as developmental delay, short stature, brachydactyly, subcutaneous ossifications, pseudohypoparathyroidism (hypocalcemia, resistance to parathormone), and resistance to thyrotropin (elevated thyroid stimulating hormone with normal or low-free thyroxine)
[44][57].
-
4. Correlation between Low Birth Weight, Obesity, and CKD
Several early-life factors may significantly influence how obesity impacts kidney health. Researchers have explored the connection between low birth weight (LBW) and susceptibility to renal disease.
A study by Hoy et al. conducted among Aborigines in Australia’s Northern Territory, where there has been a notable epidemic of renal failure
[45][58], revealed intriguing findings. Briefly, increasing BMI and decreasing birth weight act in concert to amplify the risk for albuminuria. In addition, LBW was associated with metabolic syndrome predisposition, as previously evidenced by the Hertfordshire study
[46][59].
Preterm birth presents another critical factor in this context. It not only correlates with an increased risk of obesity development but also with reduced numbers of nephrons, which can subsequently accelerate kidney deterioration. This occurs because high body weight exerts hemodynamic and metabolic effects on each nephron, and the total number of nephrons is determined at birth
[43][56]. The main determinants predisposing to reduced renal development appear to be caloric and protein malnutrition, placental malfunction, and maternal hyperglycemia
[43][56]. Notably, obese children born prematurely with focal glomerular sclerosis (FSGS) face a higher risk of progression to ESRD during childhood in comparison with their obese counterparts with FSGS born at full term. It appears that preterm birth and obesity may interact, resulting in an additive risk for the progression of kidney disease in childhood
[47][60].
Data from the CKD in Children (CKiD) study, an observational study of a large cohort of children and adolescents with mild to moderate CKD, noted a significantly higher prevalence of children who were small for gestational age and premature children compared with the general population
[48][61]. There is evidence that prematurity and LBW are risk factors not only for the development of obesity but also for hypertension and CKD in children and adults
[49][62].
Understanding this intricate relationship between LBW, obesity, and kidney health is essential for more targeted interventions and treatments to mitigate the risk of CKD and its progression in children and adolescents.
5. Neurohormonal, Metabolic and Immunological Effects of Obesity on Kidney Function
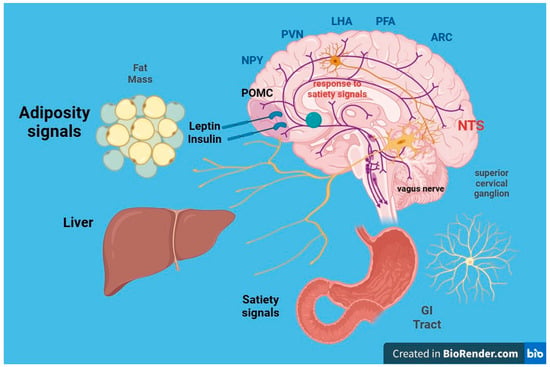
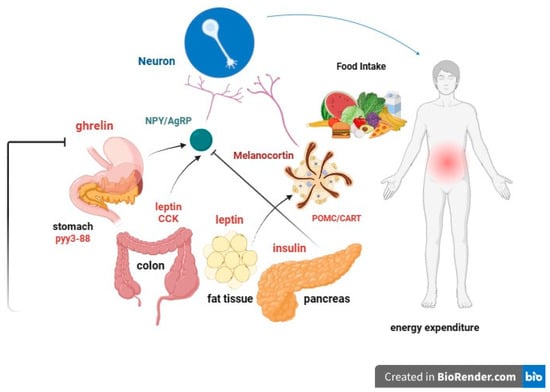
The intricate neurohormonal mechanisms responsible for obesity have been subjects of extensive research for years (
Figure 2). There is ongoing debate about whether obesity and its associated metabolic syndrome directly induce kidney damage. Although causality remains to be confirmed by cause-and-effect studies, a growing body of epidemiological studies and clinical observations suggests that the metabolic syndrome associated with obesity can indeed play a key role in the development of CKD
[50][63]. Otherwise, CKD leads to many endocrine and immunological abnormalities in adipose tissue.
Figure 2. Leptin is a peptide that acts through its own receptors with intrinsic tyrosine kinase activity expressed in the arcuate nucleus of the hypothalamus. It causes the following effects: (a) Reduction in food intake and fat mass; (b) increase in energy expenditure. The leptin signal synergizes with that of insulin. Within the arcuate nucleus of the hypothalamus, two distinct populations of neurons exist: (a) Orexigenic neurons synthesize and release neuropeptide Y (NPY) and agouti-related protein (AgRP); (b) anorexigenic neurons expressing proopiomelanocortin (POMC) and cocaine and amphetamine-related transcript (CART) synthesize and release melanocortin (MCH) and other transmitters. Leptin decreases the synthesis of NPY and AgRP in anorectic neurons and increases the synthesis of MSH in POMC neurons. From the arcuate nucleus, orexigenic and anorexigenic neurons project to the paraventricular nucleus (NPV) of the hypothalamus, regulating energy expenditure, sympathetic response, and thermoregulation.
However, despite the demonstrated correlation between obesity, metabolism, and renal insufficiency, it is crucial to note that not all overweight patients develop CKD. In fact, approximately 25% of obese individuals are metabolically healthy, implying that weight gain alone may not be the sole trigger for the development of kidney disease
[51][64].
Obesity, nonetheless, causes metabolic alterations that can contribute significantly to the onset and progression of kidney disease. Furthermore, obesity has been shown to independently influence the CKD process, even in conditions such as patients with unilateral renal agenesis
[52][65] or after unilateral nephrectomy
[53][66].
This complex scenario of multiple bidirectional interactions between adipose tissue and the kidney defines a specific adipose tissue–kidney axis.
CKD causes a redistribution of body fat with a reduction in subcutaneous fat volume and an increase in visceral and ectopic fat deposits in skeletal muscles and the liver, with a consequent harmful effect known as lipotoxicity
[54][67]. In CKD, lipid deposition also occurs in the kidney and results in increased renal inflammation
[54][67].
Additionally, it has been observed that obesity is commonly associated with a state of subclinical, low-grade inflammation in adipose tissue. Indeed, a recent study has shown that the exposure of adipose tissue to uremic serum was able to activate NF-κB (nuclear factor kappa-light-chain enhancer of activated B cells) and HIF-1α (hypoxia-inducible factor 1-alpha), resulting in adipose tissue inflammation. As evidence of the latter, adipose tissue from dialysis patients had high levels of markers of inflammation
[55][68], suggesting that it may be a source of the chronic low-grade subclinical inflammation observed in CKD patients in a manner unrelated to excess adiposity
[56][69]. CKD patients exhibit markedly higher levels of leptin, IL-6/IL-10 ratios, tumor necrosis factor, and high-sensitivity C-reactive protein than healthy individuals
[57][70]. Moreover, CKD promotes macrophage infiltration of adipose tissue, which also determines an inflammatory state and the consequent development of insulin resistance and glucose intolerance
[54][58][59][67,71,72]. It has recently been demonstrated that ESRD patients have a significantly higher density of adipose tissue macrophages compared with healthy subjects
[60][73]. Another alteration that can be observed in obesity is fibrosis of adipose tissue. The fibrotic milieu of the extracellular matrix also contributes to the inflammatory events that accompany adipocyte hypertrophy
[61][62][74,75]. There are several matrix metalloproteinases (MMPs) that regulate extracellular matrix remodeling and inflammation in adipose tissue, and among them, MMP-12 is related to renal dysfunction
[63][76].
Adipose tissue, now recognized as an endocrine organ, plays a pivotal role in these metabolic effects. It secretes various cytokines called adipokines, which play a critical role in the development of obesity complications
[64][77]. These adipokines have direct implications for lipid metabolism, inflammation, immune response, insulin resistance, atherosclerosis, metabolic homeostasis, and cell migration and proliferation
[65][78]. More than 600 proteins secreted by adipocytes have been recognized, including tumor necrosis factor, IL-6, fatty acid binding protein 4, chemerin, angiotensinogen, adiponectin, and leptin
[66][67][68][79,80,81]. The accumulation of urea in CKD also determines an increase in oxidative stress in adipose tissue, which results in an increase in the production of some adipokines such as resistin and retinol-binding protein-4, which contribute to the onset of insulin resistance
[59][72].
Cytokines produced by the adipose tissue in obese patients can induce both adaptive and maladaptive responses in renal cells, potentially compromising glomerular function through hyperfiltration
[64][77]. Several synthesized adipokines, including but not limited to leptin, adiponectin, vascular endothelial growth factor, angiopoietins, and resistin, play crucial roles in extracellular matrix accumulation, ultimately culminating in renal fibrosis
[69][82]. Other pro-inflammatory cytokines secreted by adipose tissue can activate endothelial cells and leukocytes at the level of renal microcirculation, altering endothelial barrier function and possibly leading to irreversible tubular damage with nephron loss. Notably, tumor necrosis factor-α and IL-6 have been found to be associated with CKD
[70][83]. Adipokines also indirectly damage the kidney by contributing to the development of insulin resistance and hypertension
[71][84].
Recognition of adipose tissue mediators that cause kidney damage has a relevant clinical implication as it could allow the identification of new targets for pharmacotherapies and the conception of models for predicting the risk of CKD/ESRD in obese subjects.
LEPTIN (adipose tissue) and INSULIN (pancreas) act as long-term signals of energy reserve, while GRELIN and other peptides act as short-term signals. Reductions in fat mass correlate with decreases in plasma leptin concentrations and insulin resistance. In obese individuals, circulating leptin levels are elevated and associated with leptin resistance. The leptin receptor is also expressed in the gonads, influencing sexual maturation and reproduction. Food deprivation suppresses the hypothalamic–pituitary–gonadal axis, leading to anorexia and prolonged fasting.
5.1. Insulin Resistance
Obesity is frequently accompanied by insulin resistance in peripheral tissues, resulting in hyperinsulinemia. Insulin itself exerts effects on the kidneys, either directly or indirectly, through the mediation of pro-inflammatory cytokines.
A cross-sectional analysis of data from the National Health and Nutrition Examination Survey (1999–2004), involving 2515 adolescents aged 12 to 19, unveiled noteworthy insights. Among overweight adolescents, as opposed to those adolescents with normal weight, a significant association was observed between microalbuminuria and impaired fasting glucose, highlighting the link between insulin resistance and CKD in this population
[72][85].
5.2. Leptin
Leptin, a hormone primarily metabolized in the kidneys, exerts its effect by binding to the megalin endocytic multiligand receptor in the renal proximal tubule
[73][86]. Notably, serum levels of leptin, a proinflammatory adipokine, are approximately 5–10 times higher in obese individuals than in non-obese ones
[74][87].
Leptin’s functions extend beyond the regulation of appetite, energy expenditure, and body weight. It also impairs the immune system and may exacerbate renal dysfunction
[75][88] by inducing glomerular hypertrophy and proliferation of glomerular mesangial and endothelial cells
[76][77][89,90]. That is because leptin can act on different intrarenal signaling pathways, as glomerular endothelial cells and mesangial cells abundantly express leptin receptors. Leptin promotes the expression of pro-fibrotic genes, such as TGF-β1 (transforming growth factor-β1) and pro-inflammatory cytokines. TGF-β1 can also bind to its renal receptors and increase the expression of other pro-fibrotic factors in a positive feedback loop. Furthermore, TGF-β1 is a potent stimulator of mesangial cell proliferation. Thus, the action of these molecules can result in glomerular basement membrane thickening, mesangial matrix accumulation, and mesangial hypertrophy, determining, in turn, glomerulosclerosis and proteinuria
[78][91].
Importantly, leptin levels show strong correlations with inflammation and insulin resistance, both of which are recognized risk factors for the development of CKD
[79][80][92,93].
Finally, leptin is also considered a uremic toxin that could contribute to the onset of several complications of CKD, such as cachexia, protein-energy wasting, hypertension, cardiovascular disease, and bone diseases
[81][94].
5.3. Adiponectin
Adiponectin is regarded as a predictor of chronic renal failure, and its role in safeguarding against metabolic complications associated with obesity is notable. This anti-inflammatory adipokine plays a protective role in mitigating insulin resistance and lipid accumulation
[82][95]. It also has inflammatory and anti-apoptotic properties, probably through the activation of AMP-activated protein kinase signaling
[78][83][91,96]. Interestingly, it was observed that adiponectin knockout mice develop microalbuminuria, kidney fibrosis, oxidative stress, inflammation, and podocyte dysfunction, whereas restoration of adiponectin levels rescues renal function
[84][97].
Surprisingly, levels of adiponectin are increased in patients with renal impairment. Indeed, circulating adiponectin levels exhibit an inverse relationship with both the percentage of body fat
[85][86][98,99] and the estimated GFR value
[87][100]. Furthermore, low plasma adiponectin levels are inversely correlated with proteinuria in obese subjects and may predict adverse renal outcomes in patients with type 2 DM
[88][89][101,102].
5.4. Other Adipokines
Visfatin and resistin display pro-inflammatory and atherogenic effects. Visfatin promotes the expression of TGF-β1, plasminogen activator inhibitor-1, and type I collagen, having an important pro-fibrotic role, whereas resistin stimulates the production of ICAM-1 (intercellular adhesion molecule-1) and VCAM-I (vascular cell adhesion molecule-1) and promotes the activation of the sympathetic renal system. Levels of these adipokines are increased in obesity and CKD and are associated with reduced GFR
[78][91]. Adipose tissue also produces components of the renin–angiotensin–aldosterone system (RAAS). Adipocyte-specific deletion of angiotensinogen in mice reduces plasma levels sufficiently to decrease systolic BP. Moreover, adipocytes synthesize and secrete aldosterone, which is increased in obese animals
[90][103]. Increased RAAS activation, in conjunction with glomerulomegaly and impaired sodium/glucose reabsorption, results in hypertension and glomerular hyperfiltration
[91][104].
6. Hypertension and Obesity in Children
Hypertension associated with obesity is a complex multifactorial disease, including activation of the RAAS, impaired vascular function, and increased sympathetic nervous system (SNS) activity
[92][105]. While a well-established relationship exists between obesity and hypertension in adults, the prevalence of hypertension and pre-hypertension in the pediatric setting is significantly higher among obese children and adolescents
[93][94][106,107]. The kidney is both a cause and victim of hypertension
[95][108] because, on the one hand, the presence of reduced insulin sensitivity, dysglycemia, and dyslipidemia (both precursors of hypertension) can lead to renal damage, and on the other hand, functional and structural alterations in the nephrons can contribute to increased BP
[96][109].
Hypertension is not only a risk factor for the progression of renal disease in the pediatric population with CKD
[97][110], but it is also a significant contributor to renal dysfunction in obese patients, although it is not the sole hemodynamic cause
[14][30]. Studies have shed light on the profound impact of adiposity on BP levels in the pediatric population. For instance, research by Tu et al.
[98][111] revealed a four-fold increase in BP levels across all age groups in small patients with a BMI > 85th percentile. Similarly, Sorof et al.
[99][112] demonstrated a progressive increase in hypertension prevalence in school-aged children as BMI exceeded the 95th percentile.
Furthermore, an Italian study showed that prehypertensive children exhibited reduced GFR and increased proteinuria compared with normotensive children
[100][113]. High-normal BP levels in the pre-hypertension group correlated with low-normal renal function, characterized by decreased GFR and increased proteinuria levels, albeit still within accepted normal ranges. Notably, left ventricular hypertrophy is more common in children with stage 2–4 CKD and with masked or confirmed hypertension
[101][114], underlying the importance of identifying hypertension in these cases.
Masked hypertension has emerged as a particularly strong independent predictor of left ventricular hypertrophy in the pediatric population, likely attributed to the hyperactivation of the RAAS
[102][115]. The correlation between arterial hypertension and obesity in children involves several factors. These include insulin resistance, either alone or combined with hyperleptinemia, which activates the SNS, leading to vasoconstriction, reduced renal blood flow, and subsequent activation of RAAS and water and sodium retention
[103][116].
Elevated leptin levels also contribute to increased BP and SNS activation. In the pediatric population, proinflammatory cytokines, oxidative stress pathways, sleep apnea syndrome, or poor sleep quality can further exacerbate arterial stiffness and endothelial dysfunction
[104][117], increasing the risk of hypertension in obese children
[105][118]. Moreover, low serum levels of 1,25-dihydroxyvitamin D (1,25-(OH)
2D
3) have been associated with metabolic syndrome and hypertension in Caucasian children and adolescents
[106][119].
Hyperuricemia, which can result from a high fructose diet and increased uric acid production by adipose tissue in obese individuals, also represents a potential correlation mechanism between hypertension and obesity
[107][120]. Studies in Moscow have shown hyperuricemia (>8.0 mg/dL) in a higher percentage of children with hypertension compared with those with normal BP
[108][121]. Large-scale epidemiological studies are needed to further confirm these findings.
7. Renal Biomarkers in Obese Children
CKD and renal injury due to obesity are frequently asymptomatic in the early stages and difficult to recognize, especially in the pediatric population, where individuals often remain underdiagnosed for several years. Therefore, reliable biomarkers are needed to prevent potentially very serious renal complications and dramatically improve the overall management of this disease. Traditional renal biomarkers include serum creatinine (sCr), blood urea nitrogen (BUN), urinary albumin/protein, and volume excretion values. However, sCr and BUN have limitations in distinguishing renal injury from hemodynamic changes, especially in acute kidney injury. Furthermore, their levels may not rapidly change in response to damage due to the presence of a functional reserve mechanism in nephrons. This “reservoir” consists of other nephrons that can increase their own function in response to injury. As a result, sCr and BUN levels often remain within the “normal range” until substantial injury has occurred, potentially leading to irreversible loss of a significant number of nephrons. Among the many equations available for estimating GFR, it has been seen that FAS age (height-independent full-age spectrum equation), FAS height (height-dependent full-age spectrum equation), and LMR18 (adjusted-creatinine revised Lund-Malmö equation) are the preferred serum creatinine-based formulas in children with overweight or obesity
[109][122]. While measurements of GFR using methods such as sCr or iohexol clearance provide important insights into renal function, they may not closely parallel the extent of the renal lesion.
In recent years, several studies have been conducted with the aim of identifying new biomarkers for early detection of kidney injury, although most of these are used exclusively in research settings and need further validation. Some of these
(Table 1) include urinary glutamyl aminopeptidase (GluAp), urinary podocalyxin (PCX), urinary kidney injury molecule-1 (KIM-1), alpha-1-acid glycoprotein (AGP), urinary N-acetyl-beta-D-glucosaminidase (NAG), and urinary neutrophil gelatinase-associated lipocalin (NGAL)
[110][111][123,124]. Overexpression of these biomarkers was evidenced in the obese pediatric population
[112][113][125,126]. Thus, at present, there are very few human studies concerning biomarkers of early kidney damage secondary to obesity, particularly in pediatric populations.
In obese children and adolescents, diagnostic screening strategies could be considered in order to identify any early functional and/or structural kidney injury and consequently reduce the likelihood of potential complications, improving their prognosis. However, currently, there is insufficient evidence to recommend screening for kidney complications in non-diabetic and non-hypertensive children and adolescents with obesity
[114][127].
Cystatin C is another marker used for estimating GFR and is independent of muscle mass in children
[115][116][128,129]. Although some studies, such as those by Miliku et al., suggest that both eGFR derived from creatinine (eGFR
creat) and cystatin C (eGFR
cystC) blood concentrations can be influenced by factors such as BMI and body surface area, it is worth noting that eGFR
creat is more strongly affected by lean mass percentage and fat mass percentage
[117][130]. Urinary cystatin has been found to be useful in detecting early kidney injury due to obesity in children
[111][124].
An alternative biomarker for estimating renal function is rescaled serum creatinine (SCr/Q), which accounts for sex, age, or height. SCr/Q and almost all GFR estimations correlated with obesity-related comorbidities (anthropometric and metabolic variables) in 600 children with overweight and obesity without overt kidney disease
[118][131]. Nevertheless, the progression of renal dysfunction in obesity often starts with microalbuminuria, which can eventually progress to overt proteinuria. A Framingham study has shown that overweight and obese individuals were more likely to develop proteinuria when compared to healthy individuals
[119][132]. One dramatic demonstration of the impact of obesity on renal function was observed in patients who underwent unilateral nephrectomy. Among obese nephrotic patients, 92% developed proteinuria and impaired renal function over a 10-year follow-up period, whereas only 12% of non-obese subjects experienced such complications
[120][133].
As mentioned previously, microalbuminuria functions as both an early marker of CKD and a marker of renal damage in non-diabetic patients. Csernus et al., for instance, showed that elevated levels of albuminuria and β2-microglobulinuria in obese children, compared with children with a normal body weight, suggest early renal glomerular and tubular damage, resulting from obesity
[121][134]. Ferris et al. have also highlighted that microalbuminuria is strongly correlated with the severity of obesity in adults
[122][135]. The use of multiple markers in nephrology will be useful as they can provide diverse information, including insights into the site of the lesion, the presence of inflammation, and any association with systemic diseases.
Alpha-1-acid glycoprotein (AGP) is an acute-phase protein that has both pro- and anti-inflammatory actions. Medyńska et al.
[123][136] have observed that α1-AGP increased before the onset of albuminuria, suggesting that it could be a biomarker of early glomerular damage in obese children.
Lipocalin associated with neutrophilic gelatinase (NGAL) is one of the intriguing biomarkers in this context. NGAL is a 25 kDa protein associated with neutrophilic gelatinase, and it is known to be released by renal tubular cells damaged in acute kidney injury even before a decrease in GFR occurs
[124][137]. Studies have identified NGAL concentrations in serum and urine as independent predictors of CKD progression
[125][138] in patients with moderate renal disease, and it has been noted that its urinary concentrations may predict the development of CKD in obese adolescents with normal or reduced GFR
[126][139]. Furthermore, NGAL is currently employed as an early biomarker for diabetic nephropathy
[127][140]. Notably, in a study by Goknar et al., urine NGAL concentrations did not show significant differences between obese children and healthy controls
[113][126]. However, Şen et al. observed that obese children with insulin resistance had elevated urinary levels of NGAL
[128][141]. Thus, these investigations showed mixed results regarding the possible role of NGAL as a biomarker of early kidney damage due to obesity.