2. Industrial Scale Manufacturing Techniques
2.1. Hot Melt Extrusion
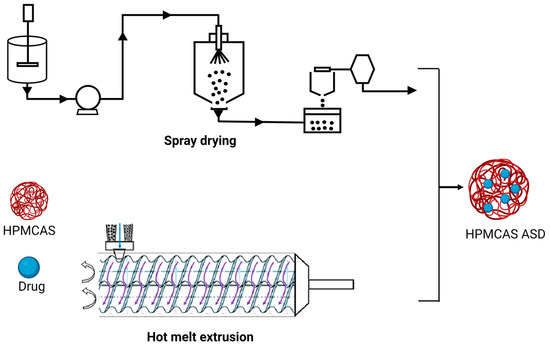
Hot melt extrusion (HME) and spray drying (SD) are the most commonly used techniques for the development of ASDs (
Figure 1). Hot melt extrusion is one of the most efficient solid dispersion manufacturing processes. The API and the polymer matrix are blended together in this procedure to create a physical combination that may be extruded under various circumstances
[37][40]. Processing parameters, for example, feed rate, shear force, temperature, die geometry, barrel design, screw speed and other variables should be taken into account when using this method
[38][41]. These parameters could have a considerable impact on the final product’s quality. HME has various advantages over other conventional approaches, including a continuous, one-step, solvent-free operation with fewer processing steps. Furthermore, it does not require compression and can increase the bioavailability as well as increase the biodistribution of the drug at the molecular level
[39][42]. However, some of the restrictions that could limit its application in scaling up and technology transfer are as follows: the need for a larger energy input as compared to other approaches, as well as the likely exclusion of some thermo-labile chemicals due to the high processing temperatures involved
[40][43]. HME has been used successfully to combine various pharmaceutical drug delivery techniques, such as conversion to salts/prodrug approach
[41][44], lipid-based delivery systems
[42][45], immediate and modified release tablets
[43][46] and 3D printing
[44][45][47,48].
Figure 1. Schematic representation of HME and SD in ASD manufacturing.
The barrel, feeder and screw parts are the major components of the extruder in HME
[46][47][49,50]. The extruder barrel is made up of a feeding portion, a venting section and a closed segment design. To soften or reduce the viscosity of the polymer, each part of the barrel can be heated
[48][51]. The feeder facilitates the transmission of material to the barrel. In the HME process, the starvation feeder with a screw speed independent of feed rate is most typically utilized. Screw elements aid in the mixing, transferring, and ultimately pushing of the melt through a die. The screw design makes it easier to set up multiple screw configurations to generate low or high shear. The conveying components aid in pushing the solid material within the barrel, whereas the kneading elements aid in the kneading process
[49][52]. In HME, the crystalline API is dissolved (when the processing temperature is above the melting point of API) or molecularly dispersed (when the processing temperature is below the melting point of API) within the molten polymer owing to the thermal and mechanical energies generated by the rotating screws and heated barrel, respectively
[50][53]. When using HME for ASD, it is necessary to ensure the processing temperature does not degrade the drug or polymer
[38][41]. The HME process can be broken down into the following steps: feeding, melting and plasticizing, conveying and mixing, venting, stripping, and downstream processing
[44][45][47,48]. While employing HME for ASD production, processing parameters of HME, such as barrel temperature, screw speed, feed rate, barrel design, die geometry and shear force, should be considered
[42][45].
2.2. Spray Drying
The spray-drying process requires numerous phases involving diverse components. The feed solution/suspension is first introduced into the drying chamber through a nozzle. Droplets are atomized when they escape the nozzle tip and come into contact with the drying fluid, which is hot gas (typically air) inside the drying chamber. The residence duration within the drying chamber is determined by the process parameters and the size of the equipment and can normally last a few milliseconds. At the dynamic droplet surface, energy/mass transfer occurs during the transit through the drying chamber. Finally, using a cyclone, the dried material is separated from the drying area and collected in a collection vessel. The exhaust gases are filtered via HEPA filters. The feed pump utilized is determined by the viscosity of the feed material and the type of atomizer system used
[51][52][53][54][55][54,55,56,57,58].
When employing this atomization setup, make sure to use a drying chamber with a large enough diameter. Material adherence to the drying chamber walls may limit its application for costly drugs and active moieties. The droplet size distribution produced by the atomization setup dictates the residence time required in the chamber as well as its dimensions. The character of the gas flow (turbulent or laminar) will also influence droplet residence time and final product moisture content. The need for strict inter-batch control of the temperature and humidity of the drying air is especially important for spray drying of amorphous systems
[54][57]. Particles are gathered after drying, utilizing certain design elements and separating mechanisms. The relatively small particle size utilized in medications necessitates this. Particle collection might occur in the bottom of the drying chamber, where it must be scrapped
[56][59]. Bag filters and cyclones are common particle separating devices. In pharmaceutical contexts, typical cyclones are reverse-flow, gas-solid separators in which centrifugal force causes the separation of two phases with differing masses
[57][60].
Spray-drying ASDs are created by evaporating a solvent or a solvent mixture from a drug and polymeric carrier solution. Atomization of the feed stock solution or suspension, droplet-gas contact, droplet drying and particle creation, and separation of dried solid particles from the drying medium (wet drying gas) are all processes in the SD process
[58][61]. To eliminate any leftover solvent, the powder may need to be dried again. While spray drying ASDs, the following critical processing variables should be considered: solution viscosity, total solid content in the spray solution, input and outlet temperatures, nozzle selection and drying gas flow rate
[59][60][61][62][62,63,64,65].
3. Marketed HPMCAS ASD Formulations
The increased Food and Drug Administration (FDA) approval of ASD products in recent years implies that ASD technology can be used to improve the dissolution rate and bioavailability of poorly soluble pharmaceuticals. Over the previous 12 years, the FDA has approved more than 20 ASD products. HPMCAS is used in eight ASD products.
3.1. Erleada®
Erleada
® is available in two strengths (60 mg and 240 mg) as an immediate-release tablet formulation of apalutamide
[63][71]. It is indicated for non-metastatic, castration-resistant prostate cancer. Apalutamide is practically insoluble at physiological pH. The preparation process includes, firstly, the preparation of the ASD of apalutamide by using HPMCAS through spray drying
[64][65][72,73]. The manufacturing process includes pre-blending of the ASD, granulation, extra-granular blending, lubrication, compression and film coating of tablets. A mean C
max of 6 µg/mL and 100 µg h/mL was achieved at steady state
[66][67][74,75].
3.2. Trikafta®
Trikafta
® is an immediate-release triple fixed-dose tablet formulation of elexacaftor, tezacaftor and ivacaftor. It is used in the treatment of cystic fibrosis in patients with F50del allele mutation. All three active ingredients have low solubility
[68][76]. Tezacaftor is a BCS Class II molecule, whereas elexacaftor and ivacaftor can be classified as either BCS Class II or IV molecules based on available information
[69][77]. Elexacaftor has a solubility of less than 0.1 mg/mL in pH buffers ranging from 1.0 to 8.0. Tezacaftor has a solubility of 0.004 mg/mL in pH 1.0, 0.003 mg/mL in pH 4.5 and 0.005 mg/mL in pH 6.8. Trikafta is prepared by a continuous process involving spray drying, blending, granulation, compression of granules into core tablets and film coating of tablets
[66][74]. A new formulation, granules, was later approved in 2021 for the same combination prepared through the same manufacturing process
[70][71][78,79].
3.3. Delstrigo®
Delstrigo
® is a fixed-dose combination tablet containing Doravirine (100 mg)/lamivudine (300 mg)/tenofovir disoproxil fumarate (300 mg) indicated for the treatment of HIV-1 infection in adult patients with no prior antiretroviral treatment history. Delstrigo
® is a bilayer tablet, comprising an ASD formulation of doravirine in the first layer, and lamivudine, tenofovir disoproxil fumarate are separately co-granulated in the second layer
[72][80]. Doravirine is a non-hygroscopic, crystalline powder, which is practically insoluble in water, considered as a Biopharmaceutical Classification System (BCS) class II compound. Lamivudine is soluble in water, sparingly soluble in methanol, slightly soluble in ethanol, and is considered a BCS class III compound. Tenofovir disoproxil fumarate is a slightly hygroscopic powder, slightly soluble in water and sparingly soluble in pH buffers 2.0–8.0. It is considered a BCS class III compound. The manufacturing process consists of spray drying, blending, lubrication, roller compaction, tablet compression and film coating. Briefly, a spray-dried intermediate (SDI) of Doravirine is manufactured by spray drying a solution of HPMCAS and doravirine dissolved in a mixture of water and organic solvent. The SDI is blended and roller compacted after combining with excipients to produce doravirine granules. Lamivudine and tenofovir disoproxil fumarate are combined with excipients, blended and roller compacted to produce the granules. Then, the granules are compressed into a bilayer tablet core and film coated
[73][81].
3.4. Symdeko®
Symdeko
® is a fixed-dose combination of tezacaftor (100 mg)/ivacaftor (150 mg) and ivacaftor (150 mg), indicated for the treatment of patients with cystic fibrosis. Tezacaftor is a crystalline material, practically insoluble in water and buffer solutions from pH 1.0 to pH 9.0. Ivacaftor, active in the combination drug product, has poor dissolution and bioavailability properties in its crystalline form; thus, preparation of an ASD using HPMCAS via spray drying was employed to overcome the solubility and dissolution-limited bioavailability
[74][82].
The oral bioavailability of tezacaftor was not determined in humans; however, it was moderate in rats and dogs (~40 to 50%). Absorption of tezacaftor does not vary during fasting or when consumed with fatty foods. In the case of ivacaftor, absorption increases three-fold with fat containing foods. The bioavailability of the crystalline drug and HPMCAS ASD dosed as oral suspension was evaluated using a vehicle composed of methyl cellulose/SLS/water (0.5/0.5/99%). The crystalline form had a bioavailability of 3–6%, whereas solid dispersion showed a bioavailability of around 100%, demonstrating that absorption was dependent on solubility
[75][83].
3.5. Orkambi®
Orkambi
® is a fixed-dose combination immediate-release tablet comprising lumacaftor (200 mg) and ivacaftor (125 mg) for the treatment of cystic fibrosis. Both lumacaftor and ivacaftor are practically insoluble in water with an aqueous solubility of 0.02 mg/mL and <0.05 mg/mL, respectively. Therefore, it is important to develop the amorphous form of active substances in order to improve solubility limited oral bioavailability. In the initial phase-I trials, the bioavailability of a capsule formulation of lumacaftor following oral dose in healthy fasted males showed a higher oral bioavailability with C
max and AUC
0-inf values 1.4 times higher compared to suspension formulation, and the median T
max values for capsule and suspension formulation were 3 h and 4 h, respectively, indicating the capsule formulation was absorbed more slowly than the suspension. Following multiple oral-dose administrations of ivacaftor in combination with lumacaftor, the exposure of ivacaftor increased approximately 2.5 to 3.4-fold when administered with food containing fat
[76][84].
3.6. Noxafil® Delayed Release Tablets
The Noxafil
® is a delayed release tablet of posaconazole, developed using HME technology to alleviate the inconsistent pharmacokinetics and variable oral bioavailability associated with the oral suspension form of posaconazole. Posaconazole is a broad-spectrum antifungal agent used for prophylaxis and the treatment of fungal infections. A pH sensitive, enteric polymer (HPMCAS) was used to prevent the release of posaconazole in the acidic gastric environment of the stomach, allowing for release in the intestinal site. This delayed release mechanism significantly improved oral bioavailability of posaconazole and achieved higher plasma drug levels with less variability for tablet ASD formulations compared to the suspension formulation. Furthermore, patient’s fed/fasting state effect or consequent administration of medications had no discernible influence on the delayed release of tablet formulation
[77][78][85,86].
3.7. Kalydeco®
Kalydeco
® is a spray-dried ASD formulation of ivacaftor, indicated for the treatment of cystic fibrosis. When administered in crystalline form, it had an oral bioavailability of 3–6% in rats due to solubility-limited oral absorption. The ASD of ivacaftor was formulated using HPMCAS to overcome the solubility limitations and improve formulation stability. The ASD formulation of ivacaftor exhibited superior solubility (67.4 μg/mL) compared to the solubility of its crystalline polymorph B (1 μg/mL). Furthermore, the ASD formulation demonstrated relative bioavailability of 100% compared to crystalline ivacaftor
[79][87]. After single-dose oral administration to healthy adult volunteers in a fed state, the mean AUC and C
max observed were 10,600 ng h/mL and 768 ng/mL, respectively
[80][81][88,89].
3.8. Zelboraf™
Zelboraf™ is the ASD formulation containing vemurafenib in HPMCAS-LF (30:70,
w/
w), produced by the solvent/antisolvent precipitation process. The process involves dissolving the drug, ionic polymer (HPMCAS) in N, N-dimethylacetamide, then the solution is precipitated by transferring it into acidified aqueous media. The precipitates are then filtered, washed repeatedly to remove the trace levels of acid and solvent content, vacuum dried and milled to obtain an amorphous powder intermediate known as microprecipitated bulk powder. This amorphous powder provides excellent physical stability with improved oral bioavailability.
The vemurafenib dissolution from its solid dispersions is ~30 times more than crystalline vemurafenib, resulting in approximately five times higher vemurafenib plasma concentrations. During phase-I clinical trials, no tumor regression was observed with conventional vemurafenib formulation at a dose as high as 1600 mg due to the limitations of poor solubility and low oral bioavailability. When patients are treated with ASD of vemurafenib, a substantial tumor regression was achieved as a result of enhanced formulation performance
[82][90].
3.9. Incivek®
Incivek
® is a tablet ASD formulation of telaprevir produced using the spray-drying technique. It is an immediate-release tablet containing 375 mg of telaprevir with a total target weight of 1 g.
Telaprevir is a hepatitis C protease inhibitor that is used to treat genotype 1 chronic hepatitis C in conjunction with peginterferon alfa and ribavirin. Telaprevir is most likely absorbed in the small intestine; nevertheless, absolute bioavailability in humans has not been determined. Telaprevir bioavailability is influenced by food. In phase-3 studies, when a 375 mg tablet formulation was administered to healthy subjects, a three- to four-fold increase in AUC and C
max of telaprevir was seen in a fed condition compared to a fasted state. The crystalline form of telaprevir has an aqueous solubility of 4.7 μg/mL and does not ionize between pH 1 and 7, suggesting the poor bioavailability of the drug. Hence, product development focused on converting the crystalline form of telaprevir to a stable amorphous form utilizing HPMCAS and spray-drying technique. The manufacturing process involves the addition of a stabilizing polymer (HPMCAS) to the spray-drying mixture containing drug and organic solvents, then secondary drying of the mixture to remove any residual solvent, tableting with extra granular excipients, compressing the tablets, and finally coating the tablets with a film coating
[83][91].