Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Fabio Zobi and Version 2 by Rita Xu.
Coordination compounds featuring one or more antifungal azole (AA) ligands constitute an interesting family of candidate molecules, given their medicinal polyvalence and the viability of drug complexation as a strategy to improve and repurpose available medications.
- repurposing
- antifungal
- azole
- metal
1. Introduction
The lack of new medically active molecules remains an issue for the treatment of several diseases and infections. In particular, cancer is responsible for approximately one death in six worldwide, and its incidence will probably raise from 18.1 million cases in 2018 to 29.4 million in 2040, according to the World Health Organization report on cancer of 2020 [1]. At the same time, antimicrobial resistance (AMR) poses a serious threat to global health, possibly causing 10 million death a year by 2050 if no efficient measures are rapidly taken [2]. Indeed, the last class of antibiotics was discovered more than 30 years ago. Since then, big pharmaceutical companies have scarcely invested in the development of new antibiotics due to an absence of financial interest [3]. Moreover, pathogens of the bacteria, fungi and protozoa groups known for developing AMR are responsible for certain neglected tropical diseases (NTDs), which are causing devastating human and material consequences in developing countries. Namely, NTDs are estimated to affect 1.7 billion people, causing 200,000 deaths each year [4].
Antifungal azoles (AAs, Scheme 1) form a class of therapeutic compounds that display selective activity against fungal pathogens by blocking ergosterol biosynthesis through the inhibition of lanosterol C14-demethylation (Figure 1A). This process occurs via coordination to the iron heme porphyrin moiety of the CYP51 enzyme (Figure 1B), which makes it impossible to perform an oxidative removal of the lanosterol methyl group [5][6][7][5,6,7]. As a consequence, the fungal organism experiences sterol depletion and accumulation of derivatives that are unable to orientate correctly within the phospholipid bilayer, ultimately leading to loss of membrane integrity and cell lysis [8][9][8,9]. Although concomitant binding to human enzymes can cause serious side effects [10], careful tuning of the drug structure and recognition of the medically relevant moieties through structure–activity relationship (SAR) studies have enabled the design of more viable generations of AAs. The resulting selectivity is quite remarkable, considering the higher similarity between humans and fungi compared to prokaryotic parasites [11].
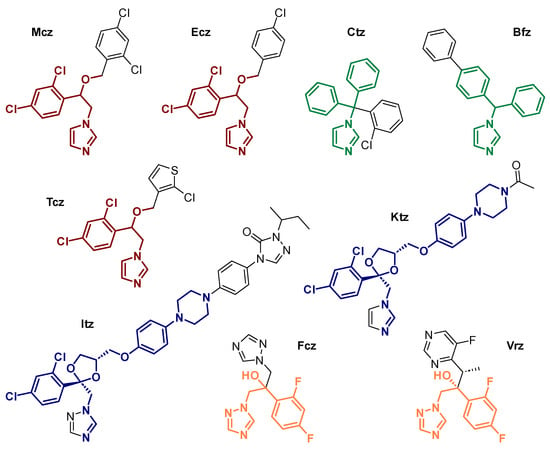
Scheme 1. Structure of selected examples of antifungal azoles (AAs): miconazole (Mcz), econazole (Ecz), tioconazole (Tcz), clotrimazole (Ctz), bifonazole (Bfz), ketoconazole (Ktz), itraconazole (Itz), fluconazole (Fcz) and voriconazole (Vrz), with emphasized structural similarities within the same family of AAs.
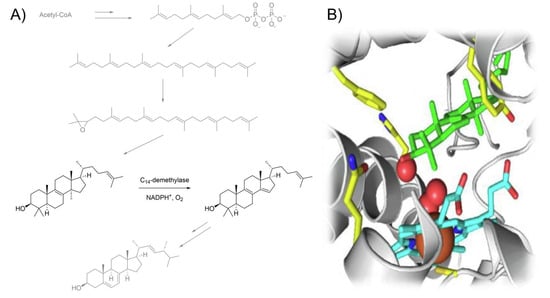
Figure 1. (A) Fungal ergosterol biosynthesis with lanosterol demethylation highlighted. (B) Schematic diagram of CYP51 catalytic site: lanosterol (green), heme (cyan), iron (orange).
Limiting AAs to their fungicidal properties would be shortsighted. Indeed, their ability to interact with CYP51 allows the inhibition of sterol biosynthesis in other organisms as well. In particular, activity against T. cruzi, the protozoan parasite causing the Chaga’s disease, and certain Leshmania species were reported, as these organisms share a similar ergosterol pathway compared to fungi [12][13][12,13].
AAs have shown their potential against prokaryotic pathogens too, as they have been known for a few decades to exhibit antibacterial activity [14][15][14,15]. In this case, the drug mechanism slightly differs from the one observed in fungal and protozoan species. Indeed, the AA probably binds the iron heme porphyrin of a bacterial deoxygenase called flavohemoglobin (Figure 2), preventing the conversion of nitrogen monoxide into nitrate by occupying the coordination site. The inability to expel NO molecules renders the bacterial organism vulnerable to NO-mediated damage induced, e.g., by host immune cells [16]. New evidence toward this mode of action was recently afforded by computational methods analyzing more than one hundred AAs and some of their close derivatives [17].
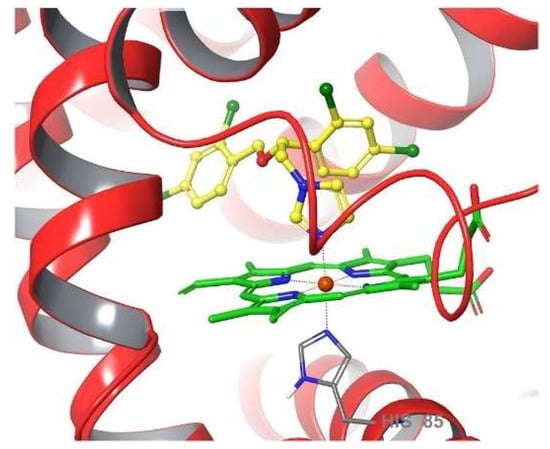
Figure 2. Schematic diagram of R. eutropha flavohemoglobin catalytic site with bound miconazole shown in yellow.
Finally, certain AAs have been studied for their anticancer properties, beginning with the investigation of ketoconazole (Ktz) as an androgen blocker through the inhibition of CYP17A1 enzyme for the treatment of prostate cancer [18]. Later, the cytotoxicity of miconazole (Mcz), econazole (Ecz), clotrimazole (Ctz) and itraconazole (Itz) was assessed in several publications, and proven to occur by various mechanisms of action including Ca2+ depletion, cell cycle arrest, glycolysis disturbance and the inhibition of the Hedgehog pathway [19]. The latter effect is especially recurrent in the case of Itz [20]. Other AAs, namely bifonazole (Bfz) [21] and tioconazole (Tcz) [22], have been scarcely analyzed for antitumoral purposes, while fluconazole (Fcz) and voriconazole (Vrz) yielded poor results in this medical application [23].
The potential combination of anticancer and antimicrobial activity in a single molecule obviously presents some advantages, considering, e.g., the necessity of prophylactic antifungal therapy in the case of immunocompromised patients who undergo certain transplants or chemotherapeutic treatments [24]. Infections caused by Candida sp. in hospitals have to be emphasized as well, because they remain a serious danger during prolonged antibiotic therapies, while multidrug treatments always carry the risk of undesired drug–drug interactions [25]. Furthermore, successfully extending the use of approved drugs outside of their original prescription constitutes a considerable gain of time and financial resources [26]. However, the repurposing of antimicrobial drugs as anticancer agents goes against the current measures preventing antibiotics misuse. It was indeed reported that such treatments promoted AMR in late stage cancer patients, threatening the health of other immunocompromised people [27].
Despite the above optimistic considerations about AAs, they are by no means new drugs and thus fail to answer the need for novel therapeutic agents. However, their Lewis basicity allows coordination to a metal center, as evidenced by their mechanisms of action. Metal complexation of well-established drugs constitutes a promising strategy to overcome loss of medication sensitivity and induced resistance [28]. Apart from the important biological activity of the metal ions, administrated in the form of approved metallodrugs [29][30][31][32][33][29,30,31,32,33], organometallic chemistry allows wider diversity in the design of medically relevant molecules, notably through higher number of possible geometries, redox features, non-covalent bonding and catalytic properties [29]. Moreover, the coordinated derivatives of biologically active ligands have been known to show synergistic effect with certain metal cores. This phenomenon, often referred to as ‘metal-drug synergism’ (MDS), can be explained by both an increase in drug activity, due to stabilization via complexation, leading to longer residence time, and a decrease in the metal toxicity, thanks to limited availability for undesired reactions compared, e.g., to the free hydrated ion [34]. In some cases, metal ions can regulate the activity of an organic drug or vice versa without requiring the intake of the corresponding metallodrug. Such examples concerning AAs are manifold in the literature [35][36][37][38][39][40][41][42][43][44][45][46][47][35,36,37,38,39,40,41,42,43,44,45,46,47].
2. Coordination Compounds of the Mcz Family of AAs
Developed by Janssen in 1968, Mcz entered the market as a topical antifungal agent in 1971. As the first synthesized and approved medically relevant AA, Mcz marked the advent of the first generation of these compounds. Simultaneously, Ecz was patented by the same company and a few years later, Tcz was designed by Pfizer [48]. Structurally speaking, these AAs are closely related to one another, as they all feature a metal-coordinating imidazole (imz), a dichlorophenyl ring and an etheric oxygen linked by an ethyl scaffold (Scheme 1). They differ from each other by the ether group bearing an aryl- or heteroarylmethyl moiety. As the predecessor of all AAs, Mcz remains the most investigated one in the field of coordination chemistry. Surprisingly, Tcz holds second place, despite Ecz displaying almost the same structure as Mcz. The first study attempting to coordinate AAs from the Mcz family to a metal center was reported by Davis et al. in 1998. The authors described the synthesis of two square planar Ir(I) complexes bearing an N-heterocyclic carbene (NHC) derivative of Mcz and Ecz (1 and 2, respectively, Scheme 2). Crystals of 2 suitable for X-ray measurements were successfully grown, which helped deducing the structure of the Mcz analog, along with NMR spectroscopy measurements. The reasoning behind this work was the production of biologically relevant organometallic NHCs known to be involved in the metabolism of vitamin B1. However, no further biological investigations were carried out [49].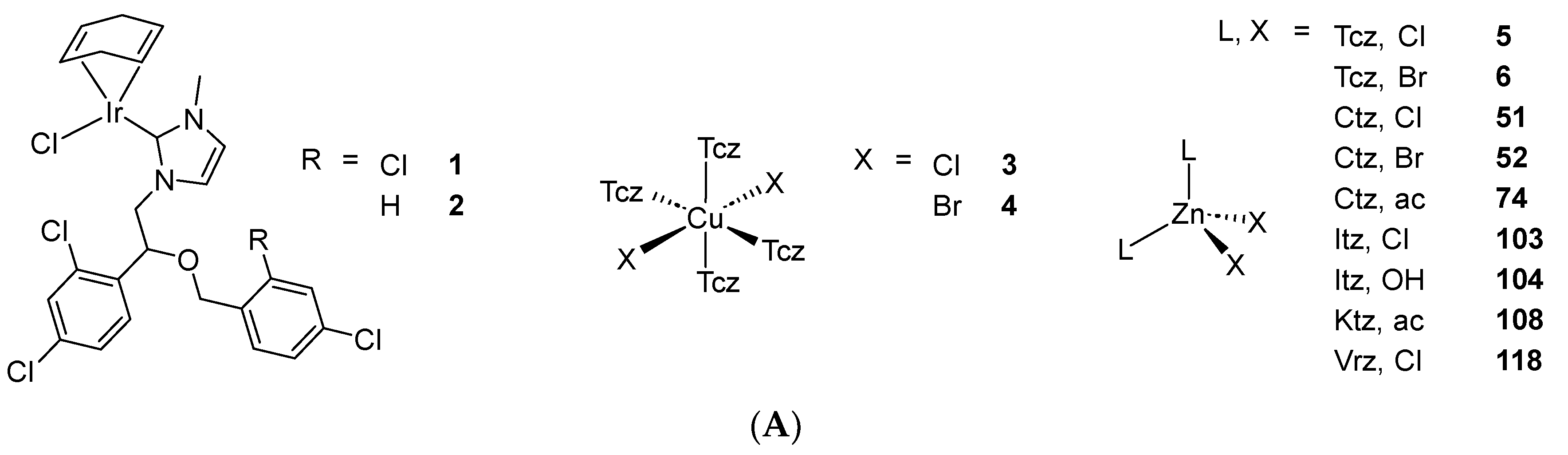
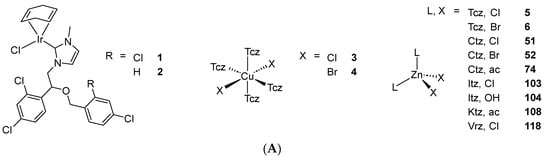
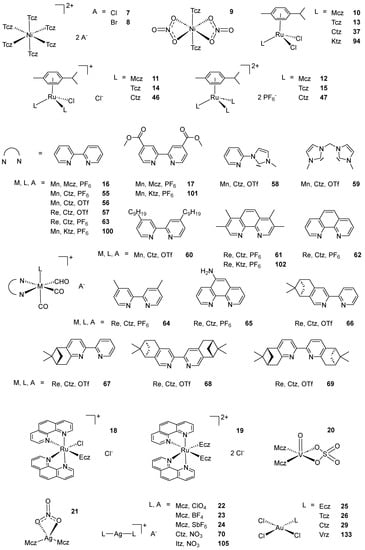
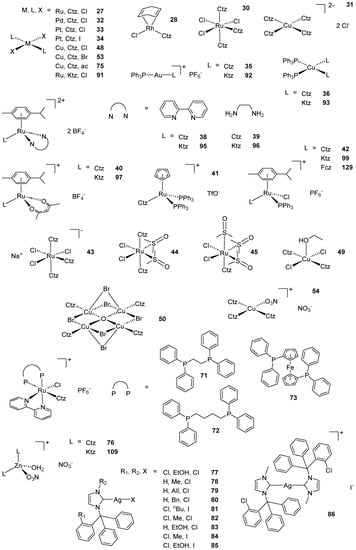
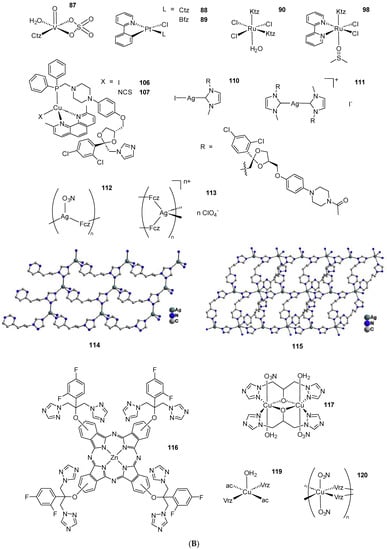
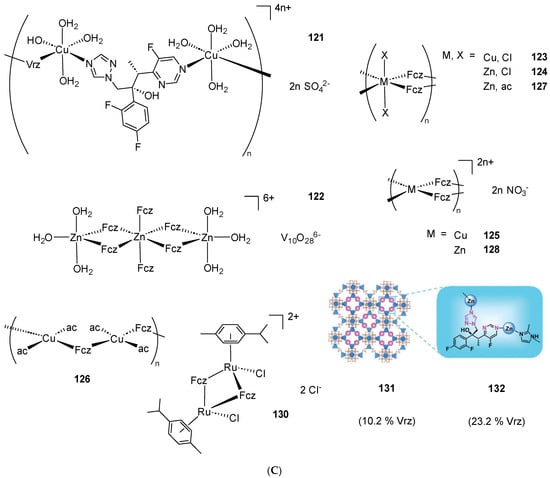
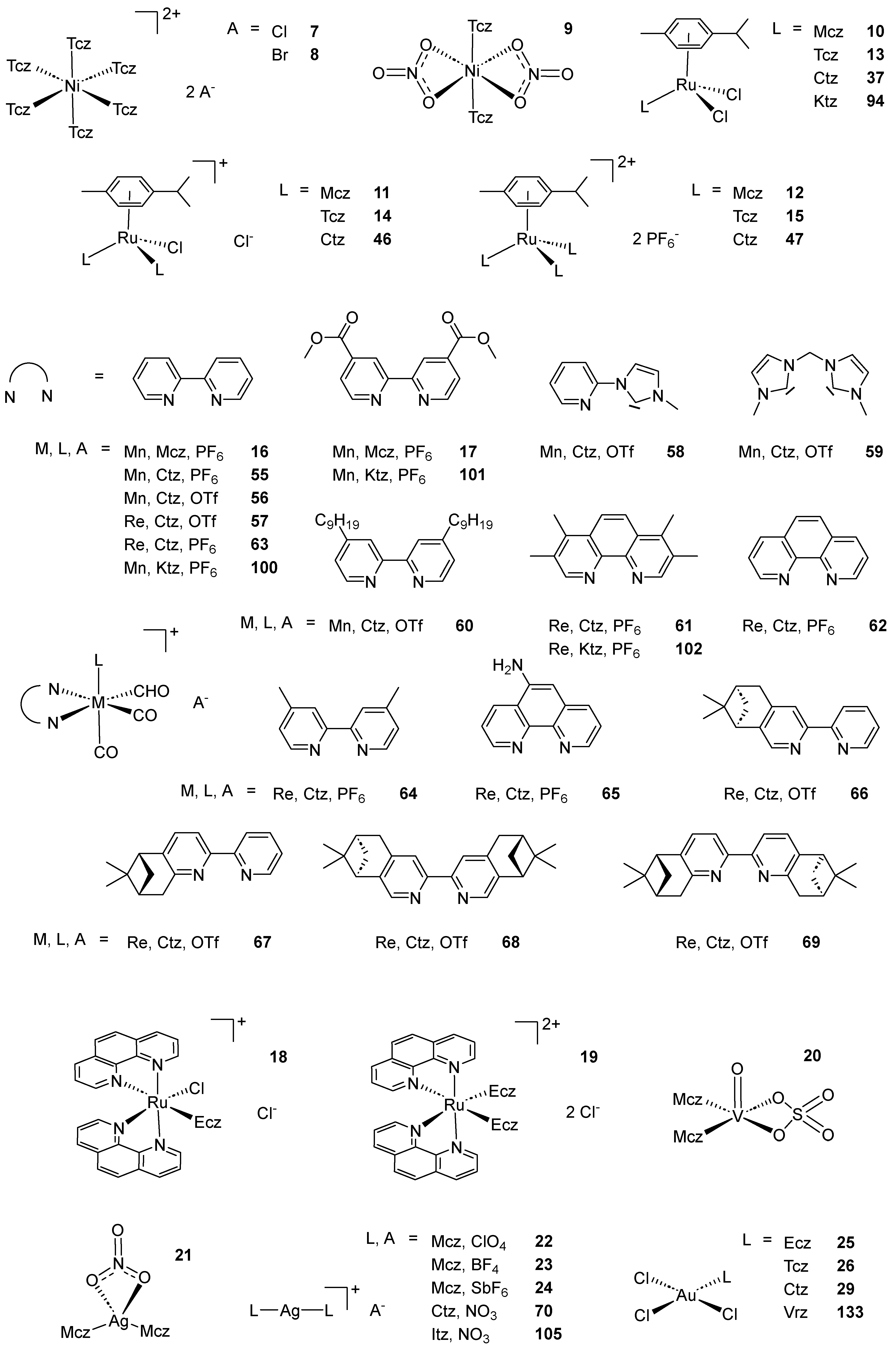
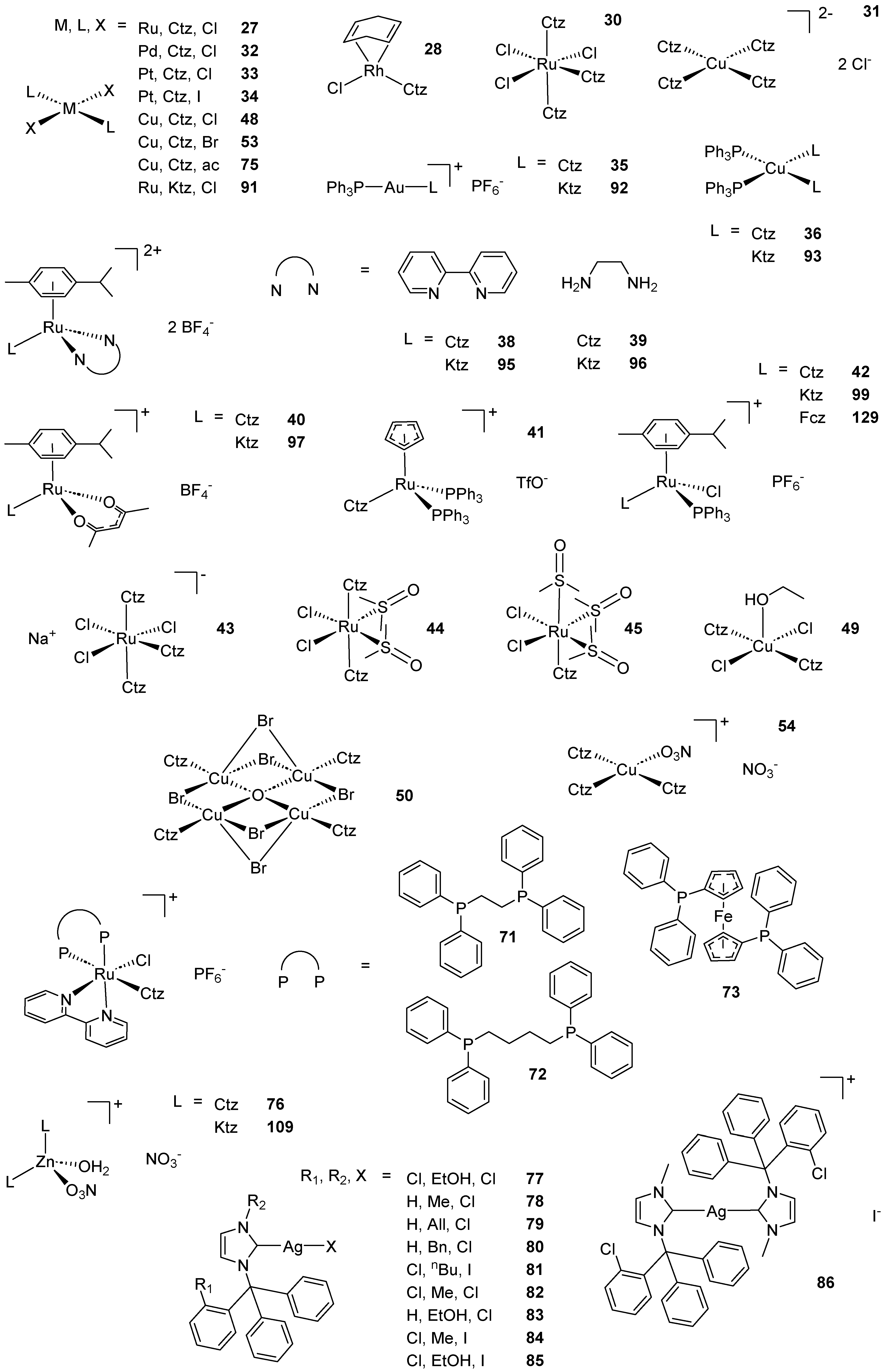
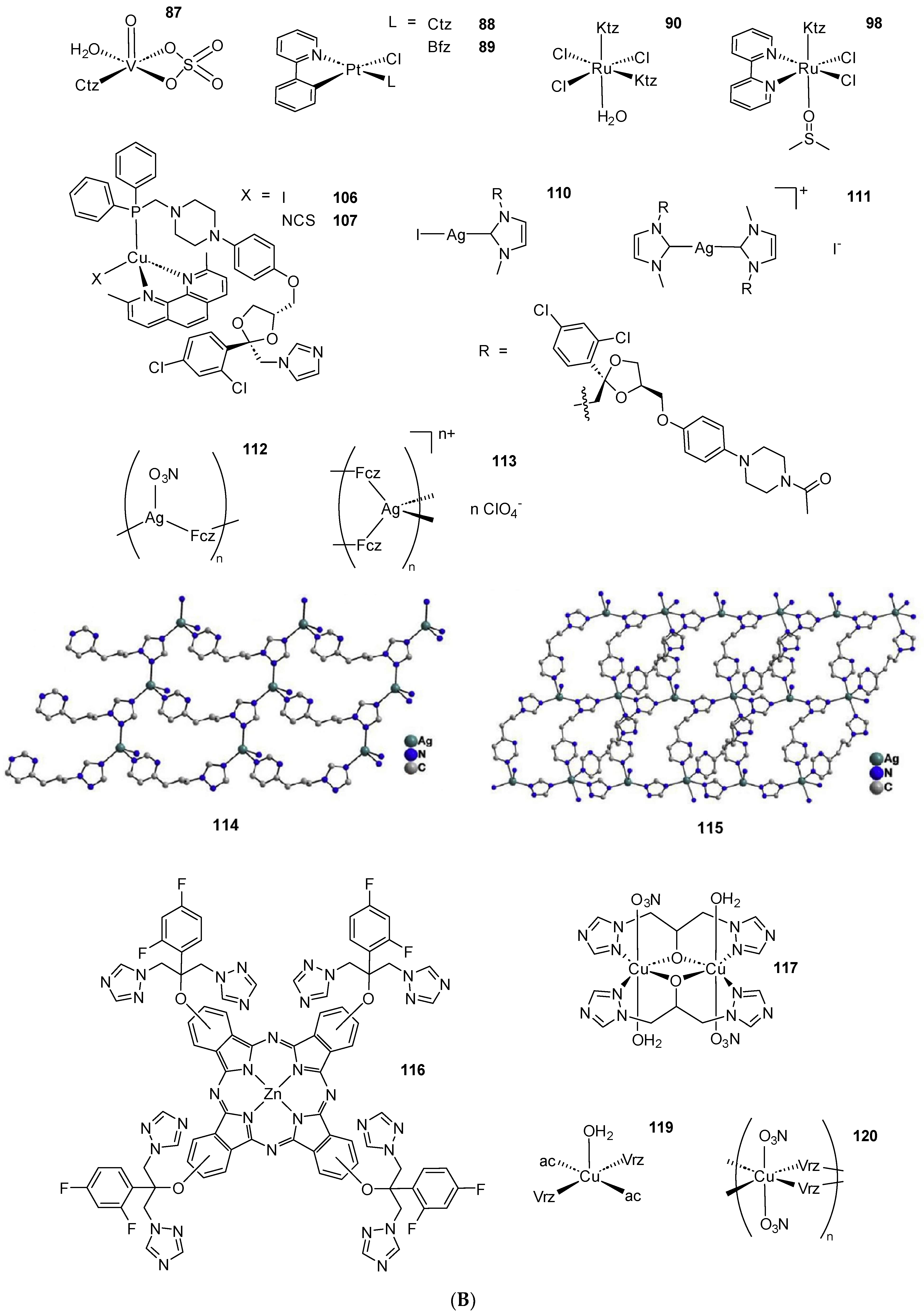

Scheme 2. (A) List of selected complexes discussed in this work (unless otherwise drawn, the coordination with AA ligands always occurs via the N3 atom of the imz ring or the N4 atom of the triazole ring); (B) the schematic view of the 2D layered structure of compounds 114 and 115 were obtained with permission; (C) the schematic representation of compounds 131 and 132 were obtained with permission.
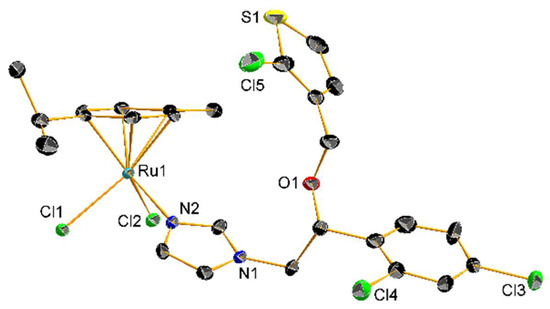
Figure 3. Crystal structure of 13 with thermal ellipsoids drawn at the 30% probability level and hydrogen atoms omitted for clarity.
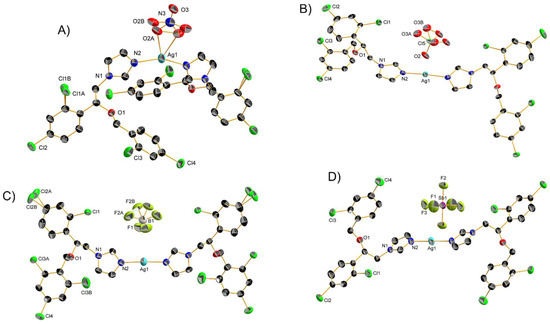
Figure 4. Crystal structures of (A) 21, (B) 22, (C) 23 and (D) 24 with thermal ellipsoids drawn at the 50% probability level and hydrogen atoms omitted for clarity.