Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Sarfaraz K. Niazi and Version 2 by Sirius Huang.
Therapeutic proteins treat many acute and chronic diseases that were, until recently, considered untreatable. However, their high development cost keeps them out of reach of most patients around the world. One possible way to make manufacturing cheaper is to use newer technologies, such as Escherichia coli to make larger molecules, like full-length antibodies, that are normally only made in Chinese Hamster Ovary (CHO) cells, switch to continuous manufacturing, and change the process to cell-free synthesis. The advantages of using E. coli include a shorter production cycle, little risk of viral contamination, cell host stability, and a highly reproducible post-translational modification.
- biosimilars
- Escherichia coli
- recombinant proteins
- monoclonal antibody
- bispecific antibody
- inclusion body
- continuous manufacturing (CM)
- cell-free protein synthesis (CFPS)
1. Introduction
Therapeutic proteins represent a diverse class of drugs first made accessible as recombinant DNA (rDNA) insulin in 1982 [1]. There are now 266 such approved proteins [2], comprising a wide range of products with unique mechanisms of action and size, ranging from peptides to monoclonal antibodies (Table 1).
Table 1.
FDA-approved therapeutic proteins as of July 2023. (
, accessed on 10 July 2023).
| Listed Protein Class | Number |
|---|---|
| Monoclonal Antibody | 94 |
| Hormone | 10 |
| Enzyme | 8 |
| Monoclonal Antibody Conjugate | 8 |
| Cytokine | 4 |
| Bispecific Antibody | 3 |
| Coagulation Factor | 3 |
| Growth Factor | 3 |
| Peptide | 3 |
| Carrier Protein | 1 |
| Enzyme Inhibitor | 1 |
| Fab | 1 |
| Fusion Proteins | 1 |
| Single-Domain Antibody | 1 |
| Toxin | 1 |
It is noteworthy that the European Medicines Agency (EMA) lists peptides as proteins, unlike the US Food and Drug Administration (FDA) [3]. Hormones, cytokines, enzymes, antibody fragments, and shorter-than-full-length antibodies that were made by E. coli were used in medicine long before Chinese Hamster Ovary (CHO) cells were used (Table 1). Some proteins still extracted from tissues can also be manufactured using E. coli. Examples include Alpha-1 antitrypsin, Antithrombin III, Botulinum toxin, C1 inhibitor, Fibrinogen, Heparin, Hirudin, Snake venom proteins, Streptokinase, Thrombin, and Urokinase. Figure 1 shows that most proteins expressed in E. coli are of lower molecular weight since most of the more complex and higher molecular weight monoclonal antibodies (mAbs) are expressed in CHO cells.
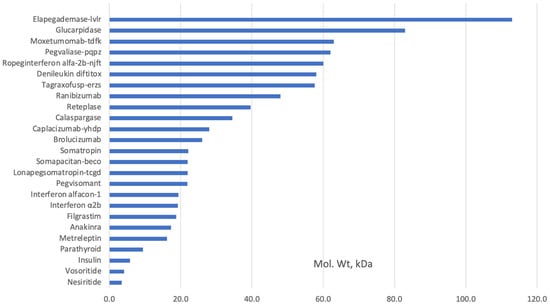
Figure 1.
Examples of FDA-approved recombinant proteins produced in
E. coli
(
, accessed on 10 July 2023).
The FDA-approved therapeutic proteins expressed in E. coli are primarily of molecular weight of less than 32 kDa; it is difficult to produce proteins higher than 100 kDa in E. coli as it places an excessive cell host load that prevents correct protein folding while maintaining adequate expression levels [4].
The value of E. coli will become more apparent when we start using it to express larger molecules, such as the mAbs. The first study on the expression of full-length (FL) immunoglobulins (FL-IgGs) in E. coli was published in 2002. Later, in 2020, a modular system-based synthetic biology technique was applied to knock down gene expression using short regulatory ribonucleic acids (RNAs) with cetuximab as a target FL-IgG for enhancing expression, reaching up to 200 mg/L [5].
Combining E. coli with emerging technologies like bioinformatics, novel methods for genetic manipulation to force E. coli to secrete heterologous proteins, and managing post-translational modification offer many new opportunities, including E. coli-based continuous manufacturing and cell-free synthesis, that can significantly reduce the cost of development and manufacturing as well as enhance product safety.
2. Background
To make recombinant DNA products, DNA from different species are joined together and then put into a host cell, usually a bacterium or mammalian cell, to make the protein that is wanted. The pioneering development of this molecular chimera dates back to 1972 [6][7][6,7] when researchers affiliated with the University of California, San Francisco, and Stanford University accomplished this technique. The United States patent for the invention was granted to Stanley Cohen from Stanford University and Herbert Boyer from the University of California, San Francisco (UCSF) in 1980. In 1976, Boyer played a crucial role in establishing Genentech, Inc. These patents have been licensed to more than 500 licensees and yielded royalties exceeding USD 250 million for Stanford and UCSF [8]. Since the FDA approved the first recombinant protein for therapeutic purposes in 1982, E. coli has remained a prominent organism for producing recombinant proteins despite the availability of many newer expression systems. Using microbial expression systems, especially E. coli, to make even heterologous recombinant proteins is still an easier and cheaper way to do it than using mammalian cell culture or other systems. E. coli presents several notable benefits in genetic manipulation, growth conditions, high product yields, product purity, absence of viral contamination, and many more.3. Bioinformatics Applications
A methodical process (Figure 2) is needed to decide if E. coli is a good system and if using it will help reach the goal of lower-cost manufacturing. The first step is to look at a number of factors, including possible splice variants, signal sequences, transmembrane helices, and post-translational modifications seen in the native protein. Protein databases, such as UniProt [9][36], are valuable initial bioinformatics resources, although they remain suggestive and not definitive. For example, knowing what can be produced in E. coli and what cannot is critical knowledge; cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) is an obligate dimer and requires N-glycosylation of Asn78 and Asn110 for dimerization [10][37], and this PTM cannot be made in E. coli. This will need a synthetic biology method since the only eukaryotic-like PTMs that E. coli can produce is disulfide bond formation in the periplasm [11][38].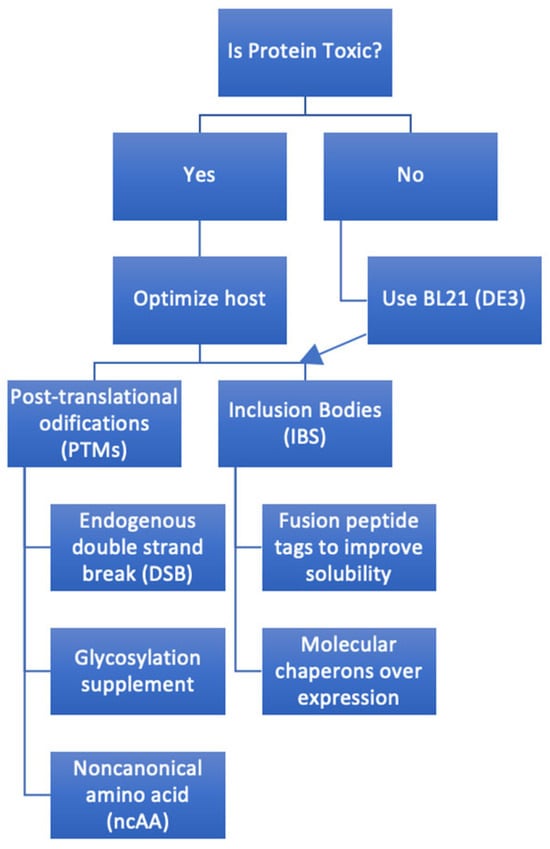
Figure 2.
A systematic process of creating a plan to express a full-length antibody in
E. coli
. Arrow indicates optimized host follow-on.
-
Exploiting the use of bioinformatics tools to determine the biophysical characteristics of the protein [14][41]. It is a complex process that involves various computational methods. These methods utilize algorithms and statistical models to analyze the protein’s primary sequence, infer its three-dimensional structure, and predict its interactions and functions.
- ○
- ○
- ○
- ○
- ○
- ○
-
- ○
- ○
- ○
- ○
- ○
4. Gene Cloning and Design
Gene cloning typically involves selecting a purification method, such as affinity chromatography, which utilizes the inherent characteristics of the protein. This can be achieved through immobilized ligand or substrate mimic chromatography, using compounds like Cibacron Blue F3GA [28][55] or cyclic peptide-based ligands [29][56]. Alternatively, a purification tag, such as a maltose-binding protein (MBP)-tag, glutathione-S-transferase (GST)-tag, or commonly a hexahistidine tag (his-tag), can be added to facilitate purification. Immobilized metal affinity chromatography (IMAC) [30][57] is commonly employed. If a protein with similar characteristics is present, its attributes can be utilized to assess the feasibility of adding a tag to the N- and C-terminus. Alternatively, one might utilize structure prediction software such as Phyre 2 [31][58]. Although N-terminal histidine tags are highly valuable and extensively employed, they can introduce heterogeneity in the final product due to varying (phospho)gluconylation occurring at the N-terminus [32][59]. After the design of the protein construct, gene design starts to yield maximum expression that depends much on cellular homeostasis, or keeping a delicate balance within the cell. When a high-copy number plasmid is employed with a robust promoter, it consistently leads to a reduced protein yield [33][60]. This is attributed to the excessive allocation of cellular resources towards synthesizing plasmid DNA and mRNA. Consequently, the abundance of mRNA exceeds the capacity of the translation machinery, resulting in suboptimal protein production. Toxic effects of overexpressed recombinant proteins on E. coli cells can be anticipated to avoid these processes [34][61]. Transcriptome analysis can identify and remove the genes in charge of the cellular stress response. The number of growth-essential genes’ down-regulated expression is reduced when cell surface receptor (CSR) is blocked [35][62]. An increasingly popular method to avoid losing plasmids during long fermentation processes is to add genes to the bacterial chromosome. However, despite their drawbacks, plasmids can be employed in their original form because they are more expeditious and cost-effective. The selection of plasmids for protein production depends on their copy quantity, which depends on the plasmid's origin of replication, promoter, and selection marker. To get the most out of the cell resources used for protein production, you need to find the right balance between the number of copies of the plasmid and the strength of the promoter, taking into account the conditions of the media. The field of synthetic biology has witnessed notable progress in developing growth-decoupled recombinant protein production. This has been achieved using the co-expression of Gp2, a peptide generated from a bacteriophage that acts as an inhibitor of RNA polymerase in Escherichia coli. This methodology facilitated the regulation of metabolic resources, ensuring their exclusive allocation towards synthesizing the intended protein. In addition to the plasmid, the origin of the gene is a crucial factor. In the past, the gene was taken directly from the living thing itself, usually by using a cDNA library made from messenger RNA (mRNA) through reverse transcription polymerase chain reaction (RT-PCR) to avoid including introns. Although the process can exhibit rapidity, cost-effectiveness, and efficiency, it can also lead to challenges associated with disparities in translation initiation and codon utilization between prokaryotic and eukaryotic organisms. Due to a significant decrease in pricing, the cost of synthesizing a gene artificially has become lower than the combined expenses of labor and materials involved in cloning a gene from a complementary DNA (cDNA) library. Synthetic genes can also alleviate the potentially harmful consequences of another dissimilarity in protein translation rates between eukaryotes and prokaryotes [36][63]. In prokaryotic organisms like Escherichia coli (E. coli), a coupling exists between the transcription and translation rates [37][64]. Specifically, transcription occurs at a rate of 50 nucleotides, whereas translation occurs at 16 amino acids.4.1. Ribosomes
In 1987 [38][65], a modified ribosome system was developed to facilitate the production of the proteins in E. coli through modifications made to the Shine–Dalgarno (SD) sequence of the mRNA and the corresponding anti-SD sequence of the 16S ribosomal RNA (rRNA). Other alternative ribosome systems can be utilized, including the orthogonal riboswitch system [39][66], the RiboTite system, and the Ribo-T system [40][41][67,68]. The riboswitch system facilitates the adjustable co-expression of several genes in a dose-dependent manner in response to tiny synthetic chemicals. On the other hand, the RiboTite system, an extension of the riboswitch technology, has demonstrated the ability to synchronize protein translation rates with protein release. The Ribo-T system utilizes a modified hybrid rRNA that combines small and large subunit rRNA sequences. This modified rRNA is connected into a single translating unit using short RNA linkers that form covalent bonds between the subunits. The functionality of the orthogonal ribosome-mRNA system has been demonstrated to sustain bacterial growth in the absence of wild-type ribosomes. Furthermore, a recent study has documented the development of an enhanced tethered version of this system [42][69].-
The characteristics and location of the ribosome binding site (RBS) and the disparities in translation rates observed in prokaryotic and eukaryotic organisms [43][70]. The ribosome binding site (RBS) plays a crucial role in translation initiation. The sequence and position of a gene relative to the initiation codon can influence translation efficiency. Customizing the RBS to the host organism might enhance the efficiency of translating the desired protein [44][71];
-
Optimization in E. coli can vary widely depending on the protein or other manufactured product. Selecting the right strain of E. coli, determining the optimal temperature, and choosing the appropriate culture media are crucial considerations for recombinant protein expression.
4.2. Promoter
Some important functional parts close to PT7 are the −35/−10 region, the translation initiation region (TIR), the operator sequence, and the TpET plasmid's replicon. There are many functional areas close to PT7, which is the core region of the pET plasmid, that control the level of expression before induction and the right transcription rate after induction. By maximizing transcription or translation levels, the T7 RNAP objective is attained. The lacUV5 promoter (PlacUV5), a strongly inducible promoter that is activated by the amino acid isopropyl-beta-d-thiogalactopyranoside (IPTG), controls this process [50][77], and the PlacUV5 is independent of recombinant product, which makes it leakier than Plac [51][78]. Three inducible promoters—ParaBAD [52][79], PrhaBAD, and Ptet—are appropriate for toxin–protein fermentation that lasts a long time. PrhaBAD and Ptet, however, more strictly control T7 RNAP transcription, giving additional expression possibilities for various recombinant products—especially dangerous proteins [53][80]. When the lac repressor gene (lacI) is altered, leaky expression is decreased by improving the ability to inhibit proteins [54][81].-
By having a mutant form of the Lac repressor protein (LacI), specifically the V192F variant, the expression of T7 RNA polymerase (RNAP) is effectively controlled to stop leakage. This mutant variant cannot bind to isopropyl β-D-1-thiogalactopyranoside (IPTG), hence preventing its activation. Consequently, the mutant LacI dynamically governs the levels of transcripts produced by T7 RNAP [56][83];
-
Because of a specific amino acid substitution (A102D), T7 RNA polymerase was less able to bind to the PT7 promoter. This changed the rate at which RNA was made. The T7 RNA polymerase (T7 RNAP) was fragmented into two segments and co-expressed with a light-responsive dimerization domain, exhibiting functional behavior upon exposure to blue light [58][85].