Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Maria Agnieszka Derkaczew and Version 2 by Lindsay Dong.
Myo-inositol belongs to one of the sugar alcohol groups known as cyclitols. Phosphatidylinositols are one of the derivatives of Myo-inositol, and constitute important mediators in many intracellular processes such as cell growth, cell differentiation, receptor recycling, cytoskeletal organization, and membrane fusion. They also have even more functions that are essential for cell survival. Mutations in genes encoding phosphatidylinositols and their derivatives can lead to many disorders.
- myo-inositol
- phosphoinositides
- phosphatidylinositol
- phosphatidylinositol phosphate
1. Introduction
Myo-inositol (MI) is the most common stereoisomer of inositol in eukaryotic cells [1]. MI was discovered by Scherer in 1850, and to this day its properties are still being investigated [2]. The physiological pool of myo-inositol is derived from diet, catabolism of phosphatidylinositols (PIs), phosphatidylinositol phosphates (PIPs)—inositol phosphates (IPs), and form various glucose-included enzymatic reactions [3][4][5][3,4,5]. The main physiological role of myoinositol stands as the precursor of the inositol phospholipids, which after modification by the hormone-stimulated inositol-phospholipid-specific phospholipase C (PLC), generate inositol 1,4,5-trisphosphate (Ins(1,4,5)P3), diacylglycerol (DAG), PI, PIP, IP, glycosylphosphatidylinositols (GPIs), Inositol trisphosphate (IP3), and inositol-phosphoglycans (IPGs) [1][3][1,3]. These molecules are used as the ubiquitous second messengers, conveying signals derived by various hormones, e.g., thyroid stimulating hormone (TSH), luteinizing hormone (LH), follicle-stimulating hormone (FSH), and insulin [1][4][6][7][1,4,6,7]. The interconversions between this group of molecules are conducted by crucial enzymes, whose dysfunction can lead to severe abnormalities, disorders, and illnesses [4][6][4,6].
2. The Family of Phosphoinositol and Phosphoinositides
Phosphatidylinositol (PtdIns), the starting point of PIP metabolism, is a ubiquitous phospholipid in eukaryotic cells present in various proportions according to the type of membrane. PIPs are all metabolized directly or sequentially from PIs [8]. The structural formulas of phosphoinositol and phosphoinositides are shown in Table 1.
Table 1.
Structural formulas of phosphoinositol and phosphoinositides.
| PI Phosphatidylinositol |
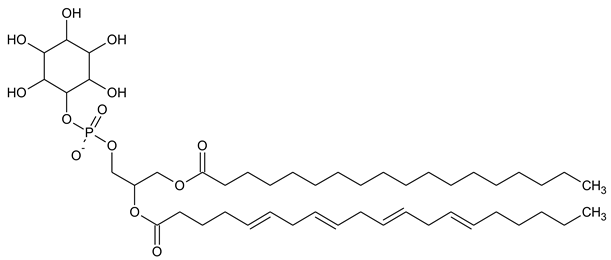 |
| PI3P Phosphatidylinositol 3-phosphate |
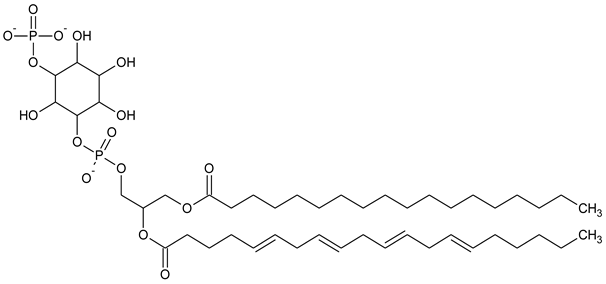 |
| PI(3,5)P2 Phosphatidylinositol 3,5-bisphosphate |
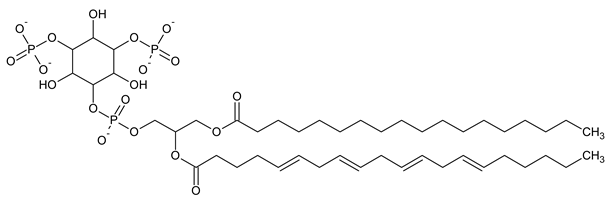 |
| PI4P Phosphatidylinositol 5-phosphate |
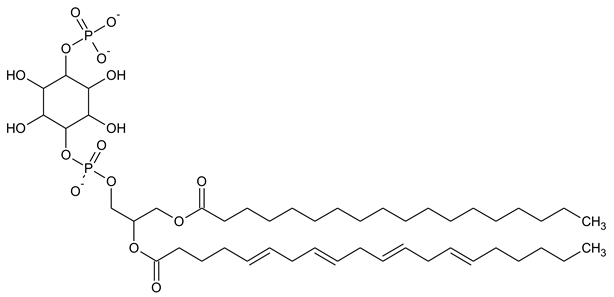 |
| PI(3,4)P2 Phosphatidylinositol 3,4-bisphosphate |
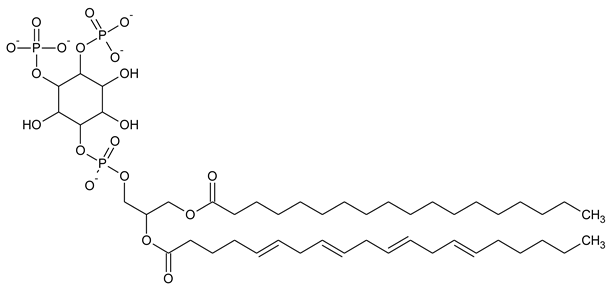 |
| PI(3,4,5)P3 Phosphatidylinositol 3,4,5-trisphosphate |
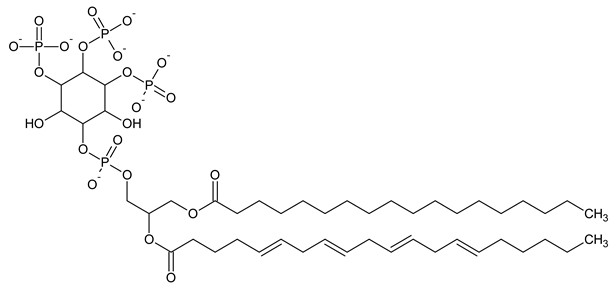 |
| PI5P Phosphatidylinositol 5-phosphate |
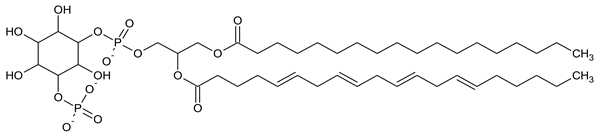 |
| PI(4,5)P2 Phosphatidylinositol 4,5-bisphosphate |
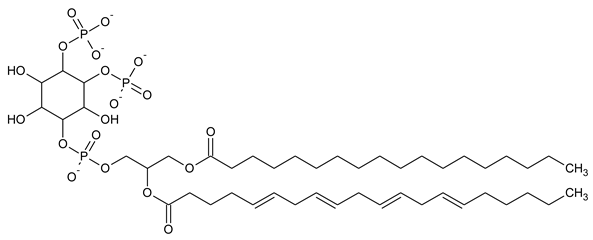 |
3. Routes and Interconversions of PIs
As previously mentioned, PI is a key compound and precursor of PIPs, which are all metabolized directly or sequentially from PI [8]. The scheme below presents detailed metabolic routes and interconversions of the PIPs family. The detailed analysis of genes and encoded enzymes is described in the table below Figure 1.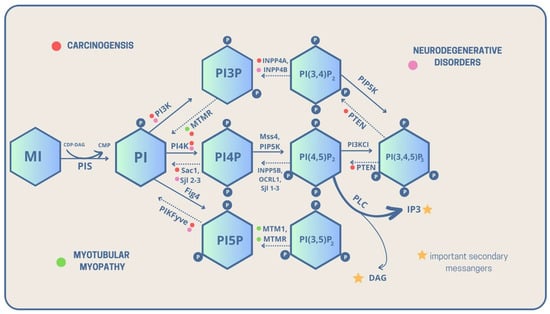
Figure 1. Metabolic interconversions of PIPs with their linkage to carcinogenesis, neurodegenerative diseases, and myotubular myopathies, with highlighted important intracellular secondary messengers [solid lines—phosphorylation, dashed lines—dephosphorylation, CDP-DAG—Cytidine diphosphate diacyloglycerol, CMP—Cytidine monophosphate, DAG—Diacylglycerol, P—phosphate group, PI—Phosphatidylinositol, PI3P—Phosphatidylinositol 3-phosphate, PI4P—Phosphatidylinositol 4-phosphate, PI5P—Phosphatidylinositol 5-phosphate, PI(3,5)P2—Phosphatidylinositol 3,5-bisphosphate, PI(3,4)P2—Phosphatidylinositol 3,4-bisphosphate, PI(4,5)P2—Phosphatidylinositol 4,5-bisphosphate, PI(3,4,5)P3—Phosphatidylinositol 3,4,5-trisphosphate, PIS—PI synthase, PLC—Phospholipase C].
PI itself is a product of the synthesis of cytidine diphosphate diacylglycerol (CPD-DAG) and MI. The reaction is conducted by PI synthase, also called phosphatidylinositol synthase 1 (PIS1) [9][10][9,10]. Then, PI phosphorylates are converted into phosphatidylinositol 3-phosphate (PI3P/PtdIns3P) [11]. The conversion is catalyzed by phosphatidylinositol 3 kinase (PI3K) and class III PI 3-kinase—vacuolar protein sorting 34 (Vps34) [12].
In the opposite direction, dephosphorylation occurs, which is conducted by PI3 phosphatases: Yeast myotubularin-related 1 (Ymr1) and Synaptojanin-like proteins 2-3 (Sjl2-3) [13][14][13,14]. Next, PI3P is phosphorylated into phosphatidylinositol 3,5-bisphosphate (PI(3,5)P2) by PI3P 5-kinase encoded by the Saccharomyces cerevisiae FAB1/PIKfyve genes [15][16][17][15,16,17].
On the other hand, dephosphorylation is conducted by Phosphoinositide 5-phosphatase—FIG4 [18]. Then, PI(3,5)P2 can transform into PtdIns5P by dephosphorylation conducted by PI3-phosphatase, MTM1/MTMR1-4, 6-8 [19]. Subsequently, phosphatidylinositol (3,4)-bisphosphate (PI(3,4)P2) can turn into PI3P by dephosphorylation catalyzed by PI4-phosphatase: phosphatidylinositol 4,5-bisphosphate 5-phosphatase A and B (INPP4A, INPP4B) [20].
Afterward, PI can also be metabolized into PtdIns4P during phosphorylation conducted by PI4-kinases: Pik1/Stt4 and PI4Kalfa/PI4Kbeta [21][22][21,22]. In dephosphorylation, enzymes such as PI4-phosphatases take part: Sjl2-3/Sac1 and SAC1 are similar to the domain of synaptojanin 1 [21][23][21,23].
Finally, PtdIns4P can turn into phosphatidylinositol 4,5-bisphosphate PI(4,5)P2 during phosphorylation conducted by PI4P 5-kinase: PIP5K α, β, and γ [24]. As for dephosphorylation, it is conducted by PI5-phosphatases: Sjl1-3, INPP5B, and OCRL1 [25][26][27][28][25,26,27,28].