Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Xiangyi Lin and Version 2 by Rita Xu.
Researchers delve into the multifaceted role of palmitoylation across various cell death modalities in the oncological context, from its intricate correlations with tumorigenesis, steered by the Asp-His-His-Cys tetrapeptide motif (DHHC) family, to the counter-process of depalmitoylation mediated by enzymes like Palmitoyl protein thioesterase-1 (PPT1).
- protein palmitoylation
- apoptosis
- autophagy
- pyroptosis
- ferroptosis
1. Introduction
Post-translational modifications (PTMs) serve as molecular conductors, orchestrating protein functions throughout cellular processes, and significantly impacting health and disease. Well-established PTMs such as phosphorylation, glycosylation, and ubiquitination have traditionally captured the focus of biomedical research [1]. Nonetheless, palmitoylation has come to be recognized as an essential contributor within this field. It plays a critical role in a variety of cellular signaling pathways, including those associated with Epidermal Growth Factor Receptor (EGFR), Ras, and Programmed Cell Death Protein 1 (PD-1)/Programmed Death-Ligand 1 (PD-L1), and is vital in metabolic regulation [2][3][2,3]. In the context of cancer, the role of palmitoylation in tumorigenesis is becoming increasingly evident. An expanding array of palmitoylated proteins has been identified as oncogenic factors, with their ability to regulate protein localization, secretion, and stability. This modulation, primarily through altered affinities between proteins and cellular membranes, underscores their potential significance in the complex mechanisms of cancer [4].
Cell death, an essential biological process, plays a pivotal role in maintaining tissue homeostasis and fostering development across diverse species. This process is intricately regulated and includes distinct modes such as apoptosis, autophagy, necroptosis, ferroptosis, and pyroptosis, each with unique morphological and biochemical signatures [5][6][5,6]. In the context of oncology, aberrant cell death pathways can initiate and promote tumorigenesis and influence therapeutic outcomes [7][8][7,8]. For example, the evasion of apoptosis is a recognized cancer hallmark, conferring a proliferative advantage to malignant cells [9]. In contrast, targeting non-apoptotic cell death pathways has emerged as a therapeutic strategy for combatting apoptosis-resistant cancers [10]. The complex relationship between cell death mechanisms and cancer highlights potential therapeutic opportunities but also presents challenges to existing paradigms, thereby driving the need for innovative research approaches [11]. An enhanced comprehension of these dynamics holds the promise of improving therapeutic strategies and broadening the spectrum of interventions against cancer [12][13][12,13].
2. Palmitoylation in Tumor Apoptosis
In the intrinsic apoptosis pathway, the tumor suppressor p53 plays a crucial role in halting the cell cycle at the G1/S checkpoint through selective DNA binding in the late G1 phase [14][82]. Decreased expression of ZDHHC16 in glioblastomas, commonly characterized by EGFR amplification, leads to p53 activation and subsequent cell cycle arrest at the G1/S checkpoint [15][16][60,61]. Additionally, p53 promotes gliomagenesis by enhancing the self-renewal and tumorigenic potential of glioma stem-like cells. This is achieved through the upregulation of ZDHHC5 transcription and the modification of EZH2 palmitoylation [17][52]. ZDHHC5-mediated palmitoylation of EZH2 activates histone H3 Lysine 27 trimethylation (H3K27me3), suppresses miR-1275, elevates Glial Fibrillary Acidic Protein (GFAP) expression, and decreases DNA methyltransferase 3 alpha (DNMT3A) binding to the OCT4 promoter, contributing to glioma malignancy [18][83]. Furthermore, p53 is palmitoylated by ZDHHC1, which influences DNMT3A’s association with the ZDHHC1 promoter, leading to its hypermethylation and the subsequent inactivation and degradation of non-palmitoylated p53, thus facilitating tumorigenesis [19][48]. Contrastingly, the S-palmitoylation of Bcl-2-associated X protein (BAX) at Cys-126, essential for initiating apoptosis, illustrates a divergence in response between normal and cancerous cells, with the former demonstrating a more rapid apoptotic response due to increased BAX S-palmitoylation [20][85]. The extrinsic apoptotic pathway is stringently modulated by protein modifications, including palmitoylation. Palmitoylation at cysteine 199 in humans and cysteine 194 in mice is critical for CD95 function, facilitating its clustering, formation of microaggregates, and internalization, which are essential for caspase-8 activation and the induction of apoptosis [21][86]. Upon palmitoylation, CD95 moves to lipid rafts enriched with cholesterol and sphingolipids, which act as pivotal signaling platforms [22][87]. Nonetheless, this apoptotic mechanism is disrupted in certain malignancies, such as Chronic Lymphocytic Leukemia, where the upregulation of APTs-governed by microRNAs like miR-138 and miR-424-disrupts CD95 palmitoylation, hinders apoptosis, and contributes to treatment resistance [23][88]. Moreover, the functionality of the extrinsic pathway is also affected by the regulation of TNF-R1, another key death receptor. The inhibition of APT2 causes an increase in TNF-R1 palmitoylation and its stabilization on the cell surface, which decreases internalization and downstream apoptotic signaling. Conversely, APT2 knockdown boosts TNF-R1 surface presence but deters apoptosis, a resistance analogous to that observed in cancers where CD95 palmitoylation is suppressed due to APT overexpression [24][89]. In the realm of tumor suppression, p53 is integral; it upregulates RARRES3 expression, which in turn impedes the Wnt/β-catenin signaling pathway through the deacylation of its components [25][90]. This RARRES3 activity inhibits cancer cell traits like proliferation, epithelial-mesenchymal transition (EMT), and stemness in breast cancer, making the dynamic between RARRES3, p53, and Wnt/β-catenin signaling a significant focus for therapeutic strategies in oncology, revealing new insights into oncogenic mechanisms [26][91] (Figure 1).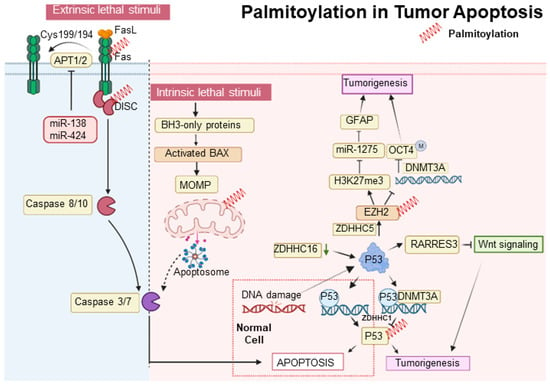
Figure 1. Palmitoylation in tumor apoptosis and cancer progression. The palmitoylation modification of proteins such as p53, BAX, and CD95 (Fas) plays a pivotal role in modulating apoptosis and influencing tumor progression. Proteins that undergo palmitoylation are denoted by a red spring.
3. Palmitoylation in Tumor Autophagy
Autophagy is frequently utilized as a mechanism to sustain cellular viability; however, it can also be associated with cell death [27][41]. It has been established that autophagy-mediated cell death and apoptosis share complex interrelationships. At the heart of this research is the AKT protein, which is crucial in regulating various cellular processes. Dysregulation of AKT has been implicated in a range of pathological conditions, particularly tumor progression due to AKT hyperactivation [28][92]. Among post-translational modifications, S-palmitoylation at the conserved residue C344 in hAKT1 is of particular interest. Alterations at this site, such as those observed in the AKT1-C344S mutant, affect AKT signaling and demonstrate a significant connection between AKT’s S-palmitoylation and cellular outcomes. Notably, this mutation alters lysosomal interactions during autophagy and reduces adipogenic differentiation potential in vitro, as depicted in Figure 1 [29][93]. Concurrently, the lysosomal enzyme PPT1, which cleaves thioester bonds in S-acylated proteins, presents intriguing consequences [30][67]. Elevated PPT1 levels are associated with increased AKT phosphorylation and enhanced cellular resistance to apoptotic stimuli. An analysis of hepatocellular carcinoma (HCC) using the TCGA dataset highlights the significance of lysosomal genes, with PPT1 positioned as a key player. By elucidating the role of PPT1 in depalmitoylation, it holds the potential to pioneer new therapeutic strategies for HCC [31][94]. In the dynamic field of oncology, the therapeutic value of PPT1 inhibitors, such as GNS561 and dimeric chloroquine (DC661), is being recognized [32][33][95,96]. DC661 surpasses hydroxychloroquine in inhibiting autophagy by targeting PPT1, while GNS561 shows potential synergy with immune checkpoint inhibitors, marking a shift in cancer treatment strategies [34][35][68,97]. Furthermore, the Phase 1b trial of GNS561 reveals its unique mechanism involving lysosomal Zn(2+) accumulation, underlining its therapeutic promise [36][98]. The influence of PPT1 extends beyond HCC. For example, in melanoma, inhibiting PPT1 enhances the efficacy of anti-PD-1 antibodies, potentially boosting T-cell mediated tumor destruction and revolutionizing melanoma therapies [37][99]. The interaction between ferroptosis and palmitoylation presents viable strategies for cancer treatment. In colorectal cancer, palmitoylation-driven lysosomal degradation of Interferon gamma receptor 1 (IFNGR1), upon optineurin depletion, impairs the IFNγ and MHC-I pathways, leading to immune evasion and intrinsic resistance to immunotherapy. Interestingly, manipulating IFNGR1 palmitoylation may intensify T-cell immune responses and improve the effectiveness of checkpoint inhibitors [38][39][100,101]. A significant finding is that the palmitoylation of the cytoplasmic domain of PD-L1 prevents its lysosomal degradation. The inhibition of this palmitoylation, or the responsible acyltransferase ZDHHC3, may strengthen antitumor immunity, offering new strategies to combat the PD-L1-mediated immune escape in cancer [40][102]. The novel compound DQ661, a dimeric quinacrine derivative, highlights the crucial role of PPT1 in lysosomal function, with implications for cancer and therapeutic resistance [41][103]. Targeted inhibition of PPT1 disrupts both the mTOR pathway and lysosomal function, signaling a new direction in cancer therapy [42][104]. Furthermore, the interaction among PPT1, TORC1, and HSP90 is becoming increasingly significant in the context of treatment resistance, indicating a potentially synergistic approach using mTOR and HSP90 inhibitors [43][105]. Inhibiting protein depalmitoylation promotes tumor cell death. A synthetic analog, DAPKA, has been shown to inhibit PPT1 in a specific fluorescence-based assay and to enhance the cytotoxicity of chemotherapeutic agents like etoposide and adriamycin in neurotumor cell lines. Overexpression of PPT1 conferred resistance to apoptosis, suggesting that its inhibition could potentiate the effects of these chemotherapeutic agents [44][106] (Figure 2).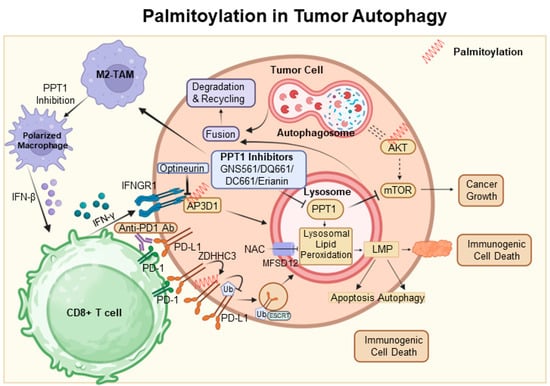
Figure 2. Palmitoylation in autophagic signaling and cancer progression. It highlights the growing importance of PPT1 in the regulation of apoptosis, autophagy, and AKT phosphorylation. Moreover, it points to the therapeutic potential of PPT1 inhibitors, such as GNS561 and DC661, in treating various cancers. The intricate relationship between PPT1 and key cellular pathways opens up new prospects for innovative cancer therapies. In this context, ZDHHC3 functions as a palmitoyltransferase, while PPT1 acts as a depalmitoylating enzyme. Proteins undergoing palmitoylation, including AKT, IFNGR1, and PD-L1, are denoted by red springs.
4. Palmitoylation in Ferroptosis and Pyroptosis
Ferroptosis is a type of cell death characterized by the accumulation of lipid peroxides, with SLC7A11 playing a crucial role in its regulation. This mechanism is particularly relevant in the oncological setting; for instance, in hepatocellular carcinoma, ferroptosis resistance—mediated by lncRNA DUXAP8 and SLC7A11—diminishes the efficacy of treatments such as sorafenib, highlighting the need for novel therapeutic strategies [45][46][107,108]. Moreover, complex interactions exist among various cell death pathways, including pyroptosis, ferroptosis, and antitumor immunity. CD8+ T cells, for example, release Granzyme A, which cleaves Gasdermin B and triggers pyroptosis. Concurrently, IFN-γ produced by CD8+ T cells suppresses SLC7A11, leading to lipid ROS accumulation and ferroptosis induction. Palmitoyltransferases ZDHHC 2, 7, 11, and 15 modify GSDME-C at Cys407 and Cys408, facilitating its separation from GSDME-N and enhancing pyroptosis [47][109]. The suppression of ZDHHC1 through promoter methylation may increase oxidative and ER stress, promoting pyroptosis via the potential substrate NLRP3, thus implicating ZDHHC1 in tumor-suppressive mechanisms [48][47]. Furthermore, erianin, noted for its anticancer effects against oral squamous cell carcinoma, inhibits cell proliferation both in vitro and in vivo. It induces a G2/M phase arrest, apoptosis, and a novel form of GSDME-mediated pyroptosis, while also affecting autophagy [49][110] (Figure 3).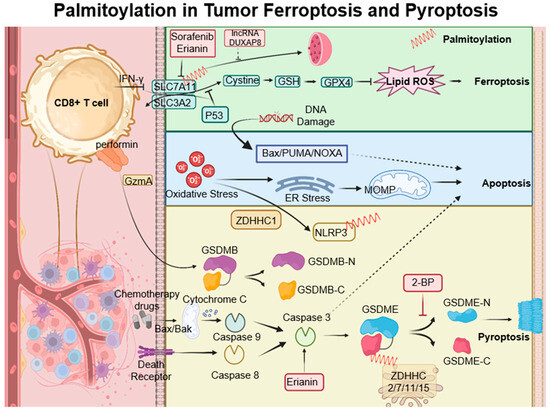
Figure 3. Palmitoylation-modulated interplay of ferroptosis and pyroptosis in oncological contexts. Ferroptosis is closely associated with critical molecules such as SLC7A11, which affect glutathione synthesis and the activity of GPX4. Palmitoylation plays a crucial role in chemically induced pyroptosis by regulating the cleavage of Gasdermin proteins, leading to inflammatory responses. CD8+ T cells influence both pyroptosis and ferroptosis via the secretion of GzmA protein and the action of IFN-γ, respectively. This sophisticated interplay between cell death pathways and immune responses opens up potential strategies for novel cancer treatments, as demonstrated by the use of therapeutic agents like erianin. ZDHHCs, which are palmitoyltransferases, modify proteins such as SLC7A11, NLRP3, and GSDME through palmitoylation, depicted as red springs.