Isolated pancreatic metastases of renal cell carcinoma (IsPMRCC) are a rare manifestation of metastatic, clear-cell renal cell carcinoma (RCC) in which distant metastases occur exclusively in the pancreas. In addition to the main symptom of the isolated occurrence of pancreatic metastases, the entity surprises with additional clinical peculiarities: (a) the unusually long interval of about 9 years between the primary RCC and the onset of pancreatic metastases; (b) multiple pancreatic metastases occurring in 36% of cases; (c) favourable treatment outcomes with a 75% 5-year survival rate; and (d) volume and growth-rate dependent risk factors generally accepted to be relevant for overall survival in metastatic surgery are insignificant in isPMRCC.
- renal cell carcinoma
- isolated pancreatic metastases
- genetics
- epigenetics
1. Introduction
2. Genetic Characteristics and Peculiarities of the isPMRCC
2.1. Clear-Cell RCC Genome
The genome of the ccRCC was deciphered as early as 2013 [57][46]. It is characterized by the biallelic absence or functional inactivation of the VHL tumour suppressor gene localized at 3p25 and the frequent inactivation of chromatin-modifying genes, such as PBRM1, BAP1 and SETD2 [58][47] (Table 1). The protein encoded by the VHL gene (pVHL) mediates its tumour-suppressive effect by binding to and mediating the proteasomal degradation of the hypoxia-inducible factor HIFα [59,60][48][49]. Under physiological conditions, HIFα subunits are unstable and are regulated by cellular oxygen content [61][50]. The loss or inactivation of VHL with consecutive inactivation of pVHL, therefore, leads to the activation and enrichment of HIF despite normoxic conditions and irrespective of the cellular oxygen availability and triggers the subsequent up-regulation of numerous HIF target genes. The activation of these HIF target genes is crucial for the formation and progression of ccRCC due to their role in promoting angiogenesis, tumour cell survival, proliferation and progression. HIFα consists of the subunits 1α and 2α, both of which are involved in ccRCC initiation [60,62][49][51]. During further ccRCC progression, however, HIF1α expression (located at chromosome 14q23 [63,64][52][53]) is lost in 30–40% since it can act as a tumour suppressor during the progression of ccRCC [60,64][49][53]. However, HIF2α acts as an oncoprotein in ccRCC. Due to the behaviour of HIF, two forms of ccRCC can be distinguished: Those in which HIF1α and 2α are overexpressed, and those in which only HIF2α is overexpressed and which are associated with enhanced cell proliferation and unfavourable prognosis [60][49]. HIF2α-triggered target factors include VEGF-α [60,65][49][54], TGF α/EGFR [66][55], c-Myc [60,67,68][49][56][57], cyclin D1 [69[58][59],70], SLC7A5-mTorC1 [60[49][60][61],71,72], GLUT1 [73,74][62][63], antioxidant enzymes [75][64], mitochondrial biogenesis factors [76][65], GAS6/tyrosine kinase AXL [77][66] and CXCR4/SDF1 [78][67], which control critical biological activities such as tumour angiogenesis, cell-autonomous proliferation, increasing glycolysis, resistance to oxidative damage, endoplasmic reticulum stress and metastatic ability [60,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81][49][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70]. Further frequently altered genes in ccRCC are chromatin-modifying genes: polybromo-1 (PBRM1), BRCA1 associated protein 1 (PAB1), SET domain containing 2 histone-lysine N-methytransferase (SETD2), located on the same 3p chromosomal region [82[71][72][73],83,84], and less frequently, lysine demethylase 5C (KDM5C) located on the X chromosome [85][74] and telomerase reverse transcriptase (TERT) promoter located on chromosome 5p [86,87][75][76]. The frequency of detectable VHL defects is estimated to be up to 90% [58,86,88,89,90][47][75][77][78][79]. In contrast, the incidence of the other altered driver genes is significantly lower: PBRM1 52.6–26.4%, SETD2 35–7.6%, BAP1 31–7.5%, KDM5C 16–3.8%, TERT 14–12.2% and mTor 13–5.7% [58,84,86,88,89,91,92,93,94][47][73][75][77][78][80][81][82][83]. It was soon recognised that these gene alterations are associated with a different tumour biology, and thus, have an influence on the course of the disease and the outcome [90,95][79][84]. PBRM1 is the most frequently mutated gene after VHL [84,92][73][81] and mutations acquired in this gene largely do not overlap with loss of function mutations in BAP1 [58,88,90,92,96][47][77][79][81][85]. PBRM1 mutations are associated with improved outcome in ccRCC [95,97][84][86] and do not correlate with decreased survival [88][77], whereas the absence of mutations of PBRM1 resulted in worse outcome [90][79]. KDM5C mutations have also been associated with improved clinical outcome in clinical reports [88,94][77][83]. In particular, the concurrent mutations of PBRM1 and KDM5C define a subgroup with increased angiogenesis associated with favourable prognosis, as Santos reports [95][84]. The similar effects of PBRM1 and KDM5C mutations on outcome are consistent with the observation that the vast majority of up- and downregulated genes after suppression of PBRM1 or KDM5C were shared [98][87]. Conversely, PAB1 mutations in ccRCC have proved to be a driver of aggressiveness and correlated with reduced outcome [58,84,88,89,90,92,99,100,101,102][47][73][77][78][79][81][88][89][90][91]. PAB1 mutations further tended to be associated with mTOR mutations [92][81]. TERT and TP53 were also identified as gene mutations associated with a poor prognosis [58,86,90,99][47][75][79][88]. However, these gene changes are generally relevant to the occurrence and course of RCC, but none of these changes can be considered specific to the occurrence of metastases, let alone isPMRCC.| Altered Genes | References | |
|---|---|---|
| Clear cell RCC | VHL Gen | [57,58,82,88][46][47][71][77] |
| Chromatin modifying genes: e.g., PBRM1, BAP1, SET2, KDM5C | [83,86,88,89,92,94][72][75][77][78][81][83] | |
| Further driver genes: e.g., pTEN, TERT, p53 | [86,89,92,93,94,99][75][78][81][82][83][88] | |
| Metastatic RCC | Loss of 9p, 14q Number of somatic copy number variants in primary ↑ metastatic potential ↑: low ITH 1 and high SCNA 2 in primary |
[63,103,104][52][92][93] |
| isPMRCC | 9p loss missing Number of somatic copy number variants ↓ chromatin-modifying genes: PBRM1 ↑, BAP1 ↓, KDM5C ↑ High genetic stability, constrained evolutionary process |
[50,91,103][80][92][94] |
2.2. Genetic Profile of Metastatic ccRCC
For the question of possible genetic characteristics of the isPMRCC, studies that specifically investigated genetic alterations that control and influence the metastatic behaviour of the RCC are therefore more relevant. Such a study was conducted and presented for the first time in 2018 by Turajlic [103][92]. In this groundbreaking analysis of 575 primary and 335 metastatic biopsies across 100 patients with metastatic ccRCC, the authors were able to identify three genetic changes that shape the metastatic behaviour of the RCC: 1. the loss of 9p21.3 and less pronounced 14q31.2 are hallmark genomic alterations at the beginning of the metastasis process; 2. the metastasis potential of RCC is reduced by low intratumoural heterogeneity and a small proportion of somatic copy-number alterations; and 3. distinct patterns of metastasis are caused by punctuated and branched evolution (Table 1).2.3. Genetic Profile of isPMRCC
So far only three publications have been presented in which this particular problem is addressed [50,91,103][80][92][94]. On the one hand, this is an inevitable consequence of the extreme rarity of isPMRCC, but on the other hand, it is also due to the fact that techniques such as next-generation sequencing have only been developed and used in recent years [82][71] (Table 1).- (a)
-
In the already cited work of Turajlic [103][92], among the 100 patients, there were also three isPMRCC observations, whose genetic profile was analysed and presented in detail for the first time. The isPMRCC showed an independent genetic profile characterized by the absence of 9p loss and a significantly lower genome instability index: Despite a 15-year and 8-year interval between primary ccRCC and clinical manifestation of PM, only one additional driver mutation was observed in two cases (mTor and SETD2, respectively) and in the third case, even after 17 years, there was no additional driver event to prove.
- (b)
-
Based on the improved prognosis of multiorgan metastases of ccRCC with concurrent PM compared to cases without PM, as shown by Grassi [105][95], and since repeatedly confirmed [11,91[11][80][86][96][97][98][99][100],97,106,107,108,109,110], Singla and colleagues in 2020 focused on the question of genetic characteristics of PM in mRCC [91][80]. (Their study group included 31 patients, but only a subgroup of just 10 (32%) met the isPMRCC criteria. However, the larger group (68%) experienced PM with simultaneous extrapancreatic multiorgan metastases of the ccRCC, which needs to be considered when assessing the relevance of the results for the specific isPMRCC topic discussed here because the detailed differences in metastasis behaviour between the two groups (single organ vs. multi-organ metastases) and the very special clinic of the isPMRCC (9.5 years metastasis-free interval until occurrence of PM and 75% 5-year survival rate, Section 1) make some genetic/epigenetic differences at least possible). In their extensive, meritorious study, Singla and colleagues were able to document genetic changes associated with less aggressive disease pathways: a low frequency of copy number variants associated with aggressiveness, such as 9p, 14q and 4q loss [63,103,104][52][92][93]. Furthermore, the authors found a low rate of PAB1 (3%) and a high rate of PBRM1 defects (77%)—changes associated with a less aggressive disease course [96,111][85][101]. Similarly, no driver mutation could be detected in TERT, which is associated with an aggressive disease course in RCC [86][75]. In contrast, KDM5C—after VHL and PBRM1—was the third most common gene mutation in the studied material with a frequency of 24%. As already pointed out above (Section 2.1), the concurrent occurrence of PBRM1 and KDM5C mutations is again a sign of a favourable course [95][84]. The high frequency of KDM5C mutations differs from metastatic ccRCC without PM in two respects. On the one hand, the value of 24% is the highest reported frequency so far [84,88,89,92][73][77][78][81]. On the other hand, in non-isPMRCC studies, KDM5C was only the fifth most common mutation [58,84,88,89,92,94][47][73][77][78][81][83]. As a further important characteristic of PMRCC, these authors also stress the unusual genetic stability of tumour cells, as limited diversification was observed both in the primary tumours leading to PM and in the subsequent PM themselves. The authors concluded that tumours and metastases from patients with PM are consistent with a constrained evolutionary process.
- (c)
-
Finally, Lou presented in 2023 an isPMRCC [50][94] that showed in the next-generation sequencing, three gene mutations (VHL, PTEN, KDM5C), a low tumour mutation burden and a microsatellite stable status. The fact that of the chromatin-modifying factors, only KDM5C was mutated is striking, as it further confirms Singla’s result of an increased frequency of KDM5C mutations (Figure 1).
-
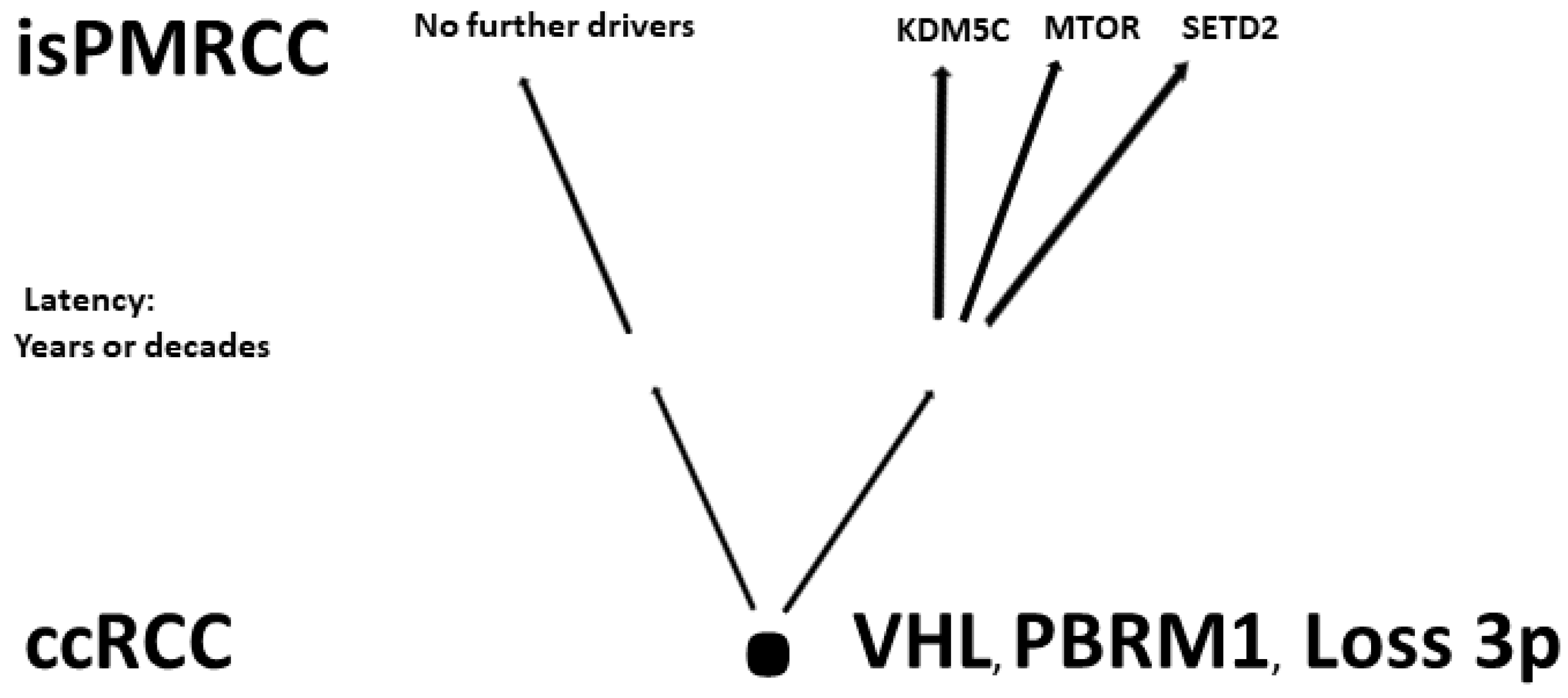 Figure 1.Involved genes in isPMRCC.
Figure 1.Involved genes in isPMRCC.
3. Conclusions
In isPMRCC, research in recent years has uncovered numerous genetic and epigenetic mechanisms that can explain the unusually protracted and favourable course and the specific response to drug therapy: e.g., high genetic stability, low frequency of copy number variants, a profile of chromatin modifying genes alterations associated with favourable course (PBRM1 ↑, PAB1 ↓) and affiliation to the angiogenetic subtype. However, the cause of the isolated occurrence of PM in isPMRCC is still unknown. The uniform long-term constant clinical course suggests at least that the phenomenon of isPMRCC is also based on uniform pathomechanisms. Therefore, genetic studies appear appropriate to clarify the mechanisms that cause the exclusive occurrence of pancreatic metastases and trigger their absence in all other organs. -
References
- Sellner, F.; Thalhammer, S.; Klimpfinger, M. Isolated pancreatic metastases of renal cell cancer: Genetics and epigenetics of an unusual tumour entity. Cancers 2022, 14, 1539.
- Moletta, L.; Friziero, A.; Serafini, S.; Grillo, V.; Pierobon, E.S.; Capovilla, G.; Valmasoni, M.; Sperti, C. Safety and efficacy of surgery for metastatic tumor to the pancreas: A single-center experience. J. Clin. Med. 2023, 12, 1171.
- Blanco-Fernández, G.; Fondevila-Campo, C.; Sanjuanbenito, A.; Fabregat-Prous, J.; Secanella-Medayo, L.; Rotellar-Sastre, F.; Pardo-Sanchez, F.; Prieto-Calvo, M.; Marin-Ortega, H.; Sanchez-Cabus, S.; et al. Pancreatic metastases from renal cell carcinoma. Postoperative outcome after surgical treatment in a Spanish multicenter study. Eur. J. Surg. Oncol. 2022, 48, 133–141.
- Malleo, G.; Salvia, R.; Maggino, L.; Marchegiani, G.; D’Angelica, M.; DeMatteo, R.; Kingham, P.; Pulvirenti, A.; Sereni, E.; Jarnagin, W.R.; et al. Long-term outcomes after surgical resection of pancreatic metastases from renal clear-cell carcinoma. Ann. Surg. Oncol. 2021, 28, 3100–3108.
- Chikhladze, S.; Lederer, A.K.; Kühlbrey, C.M.; Hipp, J.; Sick, O.; Fichtner-Feigl, S.; Wittel, U.A. Curative-intent pancreas resection for pancreatic metastases: Surgical and oncological results. Clin. Exp. Metastasis 2020, 37, 313–324.
- Di Franco, G.; Gianardi, D.; Palmeri, M.; Furbetta, N.; Guadagni, S.; Bianchini, M.; Bonari, F.; Sbrana, A.; Vasile, E.; Pollina, L.E.; et al. Pancreatic resections for metastases: A twenty-year experience from a tertiary care center. Eur. J. Surg. Oncol. 2020, 46, 825–831.
- Milanetto, A.C.; Morelli, L.; Di Franco, G.; David, A.; Campra, D.; De Paolis, P.; Pasquali, C. A plea for surgery in pancreatic metastases from renal cell carcinoma: Indications and outcome from a multicenter surgical experience. J. Clin. Med. 2020, 9, 3278.
- Anderson, B.; Williams, G.A.; Sanford, D.E.; Liu, J.; Dageforde, L.A.; Hammill, C.W.; Fields, R.C.; Hawkins, W.G.; Strasberg, S.M.; Doyle, M.B.; et al. A 22-year experience with pancreatic resection for metastatic renal cell carcinoma. HPB 2020, 22, 312–317.
- Fikatas, P.; Klein, F.; Andreou, A.; Schmuck, R.B.; Pratschke, J.; Bahra, M. Long-term survival after surgical treatment of renal cell carcinoma metastasis within the pancreas. Anticancer Res. 2016, 36, 4273–4278.
- Benhaim, R.; Oussoultzoglou, E.; Saeedi, Y.; Mouracade, P.; Bachellier, P.; Lang, H. Pancreatic metastasis from clear cell renal cell carcinoma: Outcome of an aggressive approach. Urology 2015, 85, 135–140.
- Yuasa, T.; Inoshita, N.; Saiura, A.; Yamamoto, S.; Urakami, S.; Masusa, H.; Fujii, Y.; Fukui, I.; Ishikawa, Y.; Yonese, J. Clinical outcome of patients with pancreatic metastases from renal cell cancer. BMC Cancer 2015, 15, 46.
- Schwarz, L.; Sauvanet, A.; Regenet, N.; Mabrut, J.Y.; Gigot, J.F.; Housseau, E.; Millat, B.; Ouaissi, M.; Gayet, B.; Fuks, D.; et al. Long-term survival after pancreatic resection for renal cell carcinoma metastasis. Ann. Surg. Oncol. 2014, 21, 4007–4013.
- Tosoian, J.J.; Cameron, J.L.; Allaf, M.E.; Hruban, R.H.; Nahime, C.B.; Pawlik, T.M.; Pierorazio, P.M.; Reddy, S.; Wolfgang, C.L. Resection of isolated renal cell carcinoma metastases of the pancreas: Outcomes from the Johns Hopkins Hospital. J. Gastrointest. Surg. 2014, 18, 542–548.
- Konstantinidis, I.; Dursun, A.; Zheng, H.; Wargo, J.; Thayer, S.; Castillo, C.; Warshaw, A.; Ferrone, C. Metastatic tumors in the pancreas in the modern era. J. Am. Coll. Surg. 2010, 211, 749–753.
- Bassi, C.; Butturini, G.; Falconi, M.; Sargenti, W.; Mantovavi, W.; Pederzoli, P. High recurrence rate after atypical resection for pancreatic metastases from renal cell carcinoma. Br. J. Surg. 2003, 90, 555–559.
- Ghavamian, R.; Klein, K.A.; Stephens, D.H.; Welch, T.J.; LeRoy, A.J.; Richardson, R.L.; Burch, P.A.; Zincke, H. Renal cell carcinoma metastatic to the pancreas: Clinical and radiological features. Mayo Clin. Proc. 2000, 75, 581–585.
- Thompson, L.D.; Heffess, C.S. Renal cell carcinoma to the pancreas in surgical pathology material. Cancer 2000, 89, 1076–1089.
- Yamaguchi, H.; Kimura, Y.; Nagayama, M.; Imamura, M.; Tanaka, S.; Yoshida, E.; Fujino, H.; Machiki, T.; Miyanishi, K.; Mizuguchi, T.; et al. Central pancreatectomy in portal annular pancreas for metastatic renal cell carcinoma: A case report. World J. Surg. Oncol. 2019, 17, 76.
- Bauschke, A.; Altendorf-Hofmann, A.; Deeb, A.A.; Kissler, H.; Tautenhahn, H.M.; Settmacher, U. Chirurgische Therapie von Leber und Pankreasmetastasen von Nierenzellkarzinomen. Chirurg 2021, 92, 948–954.
- Zerbi, A.; Ortolano, E.; Balzano, G.; Borri, A.; Beneduce, A.A.; Di Carlo, V. Pancreatic metastasis from renal cell carcinoma: Which patients benefit from surgical resection? Ann. Surg. Oncol. 2008, 15, 1161–1168.
- Chou, Y.; Chiou, H.; Hong, T.; Tiu, C.; Chiou, S.; Su, C.; Tsay, S. Solitary metastasis from renal cell carcinoma presenting as diffuse pancreatic enlargement. J. Clin. Ultrasound 2002, 30, 499–502.
- Sellner, F.; Thalhammer, S.; Klimpfinger, M. Isolated pancreatic metastases of renal cell carcinoma. Clinical particularities and seed and soil hypothesis. Cancers 2023, 15, 339.
- Py, J.M.; Arnaud, J.P.; Cinqualbre, J.; Adloff, M.; Bollack, C. Pancreatic metastases of nephro-epitheliomas. Apropos of 2 cases. Acta Chir. Belg. 1984, 84, 117–121.
- Strijk, S.P. Pancreatic metastases of renal cell carcinoma: Report of two cases. Gastrointest. Radiol. 1989, 14, 123–126.
- Altschuler, E.; Ray, A. Spontaneous regression of a pancreatic metastasis of a renal cell carcinoma. Arch. Fam. Med. 1998, 7, 516–517.
- Butturini, G.; Bassi, C.; Falconi, M.; Salvia, R.; Caldiron, E.; Iannucci, A.; Zamboni, G.; Grazinai, R.; Procacci, C.; Pederzoli, P.; et al. Surgical treatment of pancreatic metastases from renal cell carcinomas. Dig. Surg. 1998, 15, 241–246.
- Béchade, D.; Palazzo, I.; Desramé, J.; Duvic, C.; Hérody, M.; Didelot, F.; Coutant, G.; Algayres, J. Pancreatic metastasis of renal carcinoma: Report of three cases. Rev. Med. Interne 2002, 23, 862–866.
- Zacharoulis, D.; Asopa, V.; Karvounis, E.; Williamson, R.C. Resection of renal metastases to the pancreas: A surgical challenge. HPB 2003, 5, 137–141.
- Kapoor, R.; Kumar, R.; Dey, P.; Mittal, B.R. A late recurrence of renal cell carcinoma as pancreatic metastases: A rare disease. BMJ Case Rep. 2013, 2013, bcr2013009314.
- Chang, Y.; Liaw, C.; Chuang, C. The role of surgery in renal cell carcinoma with pancreatic metastasis. Biomed. J. 2015, 38, 173–176.
- Kusnierz, K.; Mrowiec, S.; Lampe, P. Results of surgical management of renal cell carcinoma metastatic to the pancreas. Contemp. Oncol. 2015, 19, 54–59.
- Dong, J.; Cong, L.; Zhang, T.P.; Zhao, Y.P. Pancreatic metastasis of renal cell carcinoma. Hepatobiliary Pancreat. Dis. Int. 2016, 15, 30–38.
- Ayari, Y.; Ben Rhouma, S.; Boussaffa, H.; Chelly, B.; Hamza, K.; Sellami, A.; Jrad, M.; Nouira, Y. Metachronous isolated locally advanced pancreatic metastasis from chromophobe renal cell carcinoma. Int. J. Surg. Case Rep. 2019, 60, 196–199.
- Yamawaki, M.; Takano, Y.; Noda, J.; Azami, T.; Kobayashi, T.; Niiya, F.; Maruoka, T.; Nagashama, M. A case of hemobilia caused by pancreatic metastasis of renal cell carcinoma treated with a covered metallic stent. Clin. J. Gastroenterol. 2021, 14, 905–909.
- Zhang, Z.Y.; Li, X.Y.; Bai, C.M.; Zhou, Y.; Wu, X.; Yang, A.M.; Hua, S.R. The clinicopathologic features and prognostic analysis of pancreatic metastasis from clear cell renal cell carcinoma. Zhonghua Zhong Liu Za Zhi. 2020, 42, 44–49.
- Kimura, Y.; Keira, Y.; Imamura, M.; Ito, T.; Nobuoka, T.; Mizuguchi, T.; Masumori, N.; Hasegawa, T.; Hirata, K. Histopathological aspects of pancreatic metastases in renal cell carcinoma: Does the mode of invasion permit limited resections? Pancreat. Disord. Ther. 2014, 4, 2.
- Wiltberger, G.; Bucher, J.N.; Krenzien, F.; Benzing, C.; Atanasov, G.; Schmelzle, M.; Hau, H.M.; Bartels, M. Extended resection in pancreatic metastases: Feasibility, frequency, and long-term outcome: A retrospective analysis. BMC Surg. 2015, 15, 126.
- Chatzizacharias, N.A.; Rosich-Medina, A.; Dajani, K.; Harper, S.; Huguet, E.; Liau, S.S.; Praseedom, R.K.; Jah, A. Surgical management of hepato-pancreatic metastasis from renal cell carcinoma. World J. Gastrointest. Oncol. 2017, 15, 70–77.
- Brozetti, S.; Sterpetti, A.V. Unexpected prolonged survival after extended and emergent resection of pancreatic metastases from renal cell carcinoma. J. Gastrointest. Cancer 2019, 50, 1055–1058.
- Patyutko, Y.I.; Kotelnikov, A.G.; Kriger, A.G.; Prodkuryakov, I.S.; Galkin, G.V.; Polyakov, A.N.; Fainstein, I.A. Metastatic renal cell carcinoma in the pancreas: Experience of surgical treatment. Khirurgiia 2019, 9, 25–31.
- Lauro, S.; Onesti, E.C.; Righini, R.; Carbonetti, F.; Cremona, A.; Marchetti, P. A synchronous pancreatic metastasis from renal clear cell carcinoma, with unusual CT characteristics, completely regressed after therapy with sunitinib. Case Rep. Med. 2014, 2014, 473431.
- Santoni, M.; Conti, A.; Partelli, S.; Porta, C.; Sternberg, C.N.; Procopio, G.; Bracarda, S.; Basso, U.; De Giorgi, U.; Derosa, L.; et al. Surgical resection does not improve survival in patients with renal metastases to the pancreas in the era of tyrosine kinase inhibitors. Ann. Surg. Oncol. 2015, 22, 2094–2100.
- Masetti, M.; Zanini, N.; Martuzzi, F.; Fabbri, C.; Mastrangelo, L.; Landolfo, G.; Fornelli, A.; Burzi, M.; Vezzelli, E.; Jovine, E. Analysis of prognostic factors in metastatic tumors of the pancreas: A single-center experience and review of the literature. Pancreas 2010, 39, 135–143.
- Tanis, P.J.; van der Gaag, N.A.; Busch, O.R.; van Gulik, T.M.; Gouma, D.J. Systematic review of pancreatic surgery for metastatic renal cell carcinoma. Br. J. Surg. 2009, 96, 579–592.
- Cignolli, D.; Fallara, G.; Aleotti, F.; Larcher, A.; Rosiello, G.; Rowe, I.; Basile, G.; Colandrea, G.; Martini, A.; De Cobelli, F.; et al. Pancreatic metastases after surgery for renal cell carcinoma: Survival and pathways of progression. World J. Urol. 2022, 40, 2481–2488.
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear renal cell carcinoma. Nature 2013, 499, 43–49.
- Mitchell, T.J.; Rossi, S.H.; Klatte, T.; Stewart, G.D. Genomics and clinical correlates of renal cell carcinoma. World J. Urol. 2018, 36, 1899–1911.
- Sharma, R.; Kadife, E.; Myers, M.; Kannourakis, G.; Prithviraj, P.; Ahmed, N. Determinants of resistance to VEGF-TKI and immune checkpoint inhibitors in metastatic renal cell carcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 186.
- Meléndez-Rodríguez, F.; Roche, O.; Sanchez-Prieto, R.; Aragones, J. Hypoxia-inducible factor 2-dependent pathways driving von Hippel–Lindau-Deficient renal cancer. Front. Oncol. 2018, 8, 214.
- Taylor, C.T.; Scholz, C.C. The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 2022, 18, 573–587.
- Schönenberger, D.; Harlander, S.; Rajski, M.; Jacobs, R.A.; Lundby, A.K.; Adlesic, M.; Hejhal, T.; Wild, P.J.; Lundby, C.; Frew, I.J. Formation of renal cysts and tumors in Vhl/Trp53-Deficient mice requires HIF1α and HIF2α. Cancer Res. 2016, 76, 2025–2036.
- Monzon, F.A.; Alvarez, K.; Peterson, L.; Truong, L.; Amato, R.J.; Hernandez-McClain, J.; Tannir, N.; Parwani, A.V.; Jonasch, E. Chromosome 14q loss defines a molecular subtype of clear-cell renal cell carcinoma associated with poor prognosis. Modern Pathol. 2011, 24, 1470–1479.
- Shen, C.; Beroukhim, R.; Schumacher, S.E.; Zhou, J.; Chang, M.; Signoretti, S.; Kaelin, W.G., Jr. Genetic and functional studies implicate HIF1α as a 14q kidney cancer suppressor gene. Cancer Discov. 2011, 1, 222–235.
- Kim, H.; Shim, B.Y.; Lee, S.J.; Lee, J.Y.; Lee, H.J.; Kim, I.H. Loss of Von Hippel-Lindau (VHL) tumor suppressor gene function: VHL-HIF pathway and advances in treatments for metastatic renal cell carcinoma (RCC). Int. J. Mol. Sci. 2021, 22, 9795.
- Qiu, B.; Ackerman, D.; Sanchez, D.J.; Li, B.; Ochocki, J.D.; Grazioli, A.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Brian Keith, B.; Simon, M.C. HIF-2α dependent lipid storage promotes endoplasmic reticulum homeostasis in clear cell renal cell carcinoma. Cancer Discov. 2015, 5, 652–667.
- Bailey, S.T.; Smith, A.M.; Kardos, J.; Wobker, S.E.; Wilson, H.L.; Krishnan, B.; Saito, R.; Lee, H.J.; Zhang, J.; Eaton, S.C.; et al. MYC activation cooperates with Vhl and Ink4a/Arf loss to induce clear cell renal cell carcinoma. Nat. Commun. 2017, 8, 15770.
- Gordan, J.D.; Bertovrt, J.A.; Hu, C.J.; Diehl, J.A.; Simon, M.C. HIF-2α promotes hypoxic cell proliferation by enhancing c-Myc transcriptional activity. Cancer Cell 2007, 11, 335–347.
- Latic, D.; Pejic, S.; Savic, S.; Loncar, Z.; Nikolic, I.M.; Nikolic, G.; Pavlovic, I.; Radojevic-Skodric, S. Cyclin D1 and p57 expression in relation to clinicopathological characteristics and overall survival in patients with renal cell carcinoma. JBUON 2019, 24, 301–309.
- Li, Z.; Liu, J.; Zhang, X.; Fang, L.; Zhang, C.; Zhang, Z.; Yan, L.; Tang, Y.; Fan, Y. Prognostic significance of cyclin D1 expression in renal cell carcinoma: A systematic review and meta-analysis. Pathol. Oncol. Res. 2020, 26, 1401–1409.
- Badoiu, S.C.; Greabu, M.; Miricescu, D.; Stanescu-Spinu, I.I.; Ilinca, R.; Balan, D.G.; Balcangiu-Stroescu, A.E.; Mihai, D.A.; Vacaroiu, I.A.; Stefani, C.; et al. PI3K/AKT/mTOR dysregulation and reprogramming metabolic pathways in renal cancer: Crosstalk with the VHL/HIF axis. Int. J. Mol. Sci. 2023, 24, 8391.
- Elorza, A.; Soro-Arnáiz, I.; Meléndez-Rodríguez, F.; Rodríguez-Vaello, V.; Marsboom, G.; de Cárcer, G.; Acosta-Iborra, B.; Albacete-Albacete, L.; Ordóñez, A.; Serrano-Oviedo, L.; et al. HIF2α acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol. Cell 2012, 48, 681–689.
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.H.; Harada, H. HIF-1-dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance. Int. J. Mol. Sci. 2019, 20, 238.
- Ogorevc, M.; Strikic, A.; Tomas, S.Z. Determining the immunohistochemical expression of GLUT1 in renal cell carcinoma using the HSCORE method. Biomed. Rep. 2021, 15, 79.
- Bertout, J.A.; Majmundara, A.J.; Gordana, J.D.; Lama, J.C.; Ditsworth, D.; Keitha, B.; Brown, E.J.; Nathanson, K.L.; Simon, M.C. HIF2α inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc. Natl. Acad. Sci. USA 2009, 106, 14391–14396.
- Popov, L.D. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899.
- Gustafsson, A.; Boström, A.K.; Ljungberg, B.; Axelson, H.; Dahlbäck, B. Gas6 and the receptor tyrosine kinase Axl in clear cell renal cell carcinoma. PLoS ONE 2009, 4, e7575.
- Alsayed, R.K.; Khan, A.Q.; Ahmad, F.; Ansari, A.W.; Alam, M.A.; Buddenkotte, J.; Steinhoff, M.; Uddin, S.; Ahmad, A. Epigenetic regulation of CXCR4 signalling in cancer pathogenesis and progression. Semin. Cancer Biol. 2022, 86, 697–708.
- Shinojima, T.; Oya, M.; Takayanagi, A.; Mizuno, R.; Shimizu, N.; Murai, M. Renal cancer cells lacking hypoxia inducible factor (HIF)-1a expression maintain vascular endothelial growth factor expression through HIF-2a. Carcinogenesis 2007, 28, 529–536.
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumor growth and progression. Nat. Rev. Cancer 2011, 12, 9–22.
- Gouw, A.M.; Toal, G.G.; Felsher, D.W. Metabolic vulnerabilities of MYC-induced cancer. Oncotarget 2016, 7, 29879–29880.
- Jonasch, E.; Walker, C.L.; Rathmell, W.K. Clear cell renal cell carcinoma ontogeny and mechanisms of lethality. Nat. Rev. Nephrol. 2021, 17, 245–261.
- Eckel-Passow, J.E.; Serie, D.J.; Cheville, J.C.; Ho, T.H.; Kapur, P.; Brugarolas, J.; Thompson, R.H.; Leibovich, B.C.; Kwon, E.D.; Joseph, R.W.; et al. BAP1 and PBRM1 in metastatic clear cell renal cell carcinoma: Tumor heterogeneity and concordance with paired primary tumor. BMC Urol. 2017, 17, 19.
- Hakimi, A.A.; Ostrovnaya, I.; Reva, B.; Schultz, N.; Chen, Y.B.; Gonen, M.; Liu, H.; Takeda, S.; Voss, M.H.; Satish, K.; et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: A report by MSKCC and the KIRC TCGA Research Network. Clin. Cancer Res. 2013, 19, 3259–3267.
- Zheng, Q.; Li, P.; Zhou, X.; Qiang, Y.; Fan, J.; Lin, Y.; Chen, Y.; Guo, J.; Wang, F.; Xue, H.; et al. Deficiency of the X-inactivation escaping gene KDM5C in clear cell renal cell carcinoma promotes tumorigenicity by reprogramming glycogen metabolism and inhibiting ferroptosis. Theranostics 2021, 11, 8674.
- Casuscelli, J.; Becerra, M.F.; Manley, B.J.; Zabor, E.C.; Reznik, E.; Redzematovic, A.; Arcila, M.E.; Tennenbaum, D.M.; Ghanaat, M.; Kashan, M.; et al. Characterization and impact of TERT promoter region mutations on clinical outcome in renal cell carcinoma. Eur. Urol. Focus 2019, 4, 642–649.
- Ma, R.; Liu, C.; Lu, M.; Yuan, X.; Cheng, G.; Kong, F.; Lu, J.; Strååt, K.; Björkholm, M.; Ma, L.; et al. The TERT locus genotypes of rs2736100-CC/CA and rs2736098-AA predict shorter survival in renal cell carcinoma. Urol. Oncol. 2019, 37, 301.e1–301.e10.
- Liu, Y.; Li, Y.; Xu, H.; Zhou, L.; Yang, X.; Wang, C. Exploration of morphological features of clear cell renal cell carcinoma with PBRM1, SETD2, BAP1, or KDM5C mutations. Int. J. Surg. Pathol. 2023, 1, 10.
- Carlo, M.I.; Manley, B.; Patil, S.; Woo, K.M.; Coskey, D.T.; Redzematovic, A.; Arcila, M.; Ladanyi, M.; Lee, W.; Chen, Y.B.; et al. Genomic alterations and outcomes with VEGF-targeted therapy in patients with clear cell renal cell carcinoma. Kidney Cancer 2017, 1, 49–56.
- Voss, M.H.; Reising, A.; Cheng, Y.; Patel, P.; Marker, M.; Kuo, F.; Chan, T.A.; Choueiri, T.K.; Hsieh, J.J.; Hakimi, A.A.; et al. Genomically annotated risk model for advanced renal-cell carcinoma: A retrospective cohort study. Lancet Oncol. 2018, 19, 1688–1698.
- Singla, N.; Xie, Z.; Zhang, Z.; Gao, M.; Yousuf, Q.; Onabolu, O.; McKenzie, T.; Tcheuyap, V.T.; Ma, Y.; Choi, J.; et al. Pancreatic tropism of metastatic renal cell carcinoma. JCI Insight 2020, 5, e134564.
- Brugarolas, J. Molecular genetics of clear-cell renal cell carcinoma. J. Clin. Oncol. 2014, 32, 1968–1976.
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867.
- Hsieh, J.J.; Chen, D.; Wang, P.I.; Marker, M.; Redzematovic, A.; Chen, Y.B.; Selcuklu, S.D.; Weinhold, N.; Bouvier, N.; Huberman, K.H.; et al. Genomic biomarkers of a randomized trial comparing first-line Everolimus and Sunitinib in patients with metastatic renal cell carcinoma. Eur. Urol. 2017, 71, 405–414.
- Santos, M.; Lanillos, J.; Caleiras, E.; Valdivia, C.; Roldan-Romero, J.M.; Laínez, N.; Puente, J.; Beuselinck, B.; Oudard, S.; Zucman-Rossi, J.; et al. PBRM1 and KDM5C cooperate to define high-angiogenesis tumors and increased antiangiogenic response in renal cancer. Am. J. Cancer Res. 2023, 13, 2116–2125.
- Gu, Y.F.; Cohn, S.; Christie, A.; McKenzie, T.; Wolff, N.; Do, Q.N.; Madhuranthakam, A.J.; Pedrosa, I.; Wang, T.; Dey, A.; et al. Modeling renal cell carcinoma in mice: Bap1 and Pbrm1 inactivation drive tumor grade. Cancer Discov. 2017, 7, 900–917.
- Shaya, J.A.; Lin, X.; Weise, N.; Cabal, A.; Panian, J.; Derweesh, I.H.; McKay, R.R. Prognostic significance of pancreatic metastases in patients with advanced renal cell carcinoma treated with systemic therapy. Clin. Genitourin. Cancer 2021, 19, e367–e373.
- Liao, L.; Liu, Z.Z.; Langbein, L.; Cai, W.; Cho, E.; Na, J.; Niu, X.; Jiang, W.; Zhong, Z.; Cai, W.L.; et al. Multiple tumor suppressors regulate a HIF-dependent negative feedback loop via ISGF3 in human clear cell renal cancer. eLife 2018, 7, e37925.
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018, 23, 313–326.e5.
- Da Silva, J.L.; Dos Santos, A.; Nunes, N.; Lino da Silva, F.; Ferreira, C.; de Melo, A.C. Cancer immunotherapy: The art of targeting the tumor immune microenvironment. Cancer Chemother. Pharmacol. 2019, 84, 227–240.
- Joseph, R.W.; Kapur, P.; Serie, D.J.; Eckel-Passow, J.E.; Parasramka, M.; Ho, T.; Cheville, C.J.; Frenkel, E.; Rakheja, D.; Brugarolas, J.; et al. Loss of BAP1 protein expression is an independent marker of poor prognosis in patients with low-risk clear cell renal cell carcinoma. Cancer 2014, 120, 1059–1067.
- de Cubas, A.A.; Rathmell, W.K. Epigenetic modifiers: Activities in renal cell carcinoma. Nat. Rev. Urol. 2018, 15, 599–614.
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Chambers, T.; Lopez, J.I.; Nicol, D.; O’Brien, T.; Larkin, J.; Horswell, S.; et al. Tracking cancer evolution reveals constrained routes to metastases: TRACERx Renal. Cell 2018, 173, 581–594.
- La Rochelle, J.; Klatte, T.; Dastane, A.; Rao, N.; Seligson, D.; Said, J.; Shuch, B.; Zomorodian, N.; Kabbinavar, F.; Belldegrun, A.; et al. Chromosome 9p deletions identify an aggressive phenotype of clear cell renal cell carcinoma. Cancer 2010, 116, 4696–4702.
- Lou, Y.; Guo, K.; Zheng, S. Pancreatic metastasis of renal cell carcinoma 16 years after nephrectomy. Front. Oncol. 2023, 13, 1091635.
- Grassi, P.; Verzoni, E.; Mariani, L.; De Braud, F.; Coppa, J.; Mazzaferro, V.; Procopio, G. Prognostic role of pancreatic metastases from renal cell carcinoma: Results from an Italian center. Clin. Genitourin. Cancer 2013, 11, 484–488.
- Sellner, F.; Thalhammer, S.; Klimpfinger, M. Tumour evolution and seed and soil mechanism in pancreatic metastases of renal cell carcinoma. Cancers 2021, 13, 1342.
- Kalra, S.; Atkinson, B.J.; Matrana, M.R.; Matin, S.F.; Wood, C.G.; Karam, J.A.; Tamboli, P.; Sircar, K.; Rao, P.; Corn, P.G.; et al. Prognosis of patients with metastatic renal cell carcinoma and pancreatic metastases. BJU Int. 2016, 117, 761–765.
- Chrom, P.; Stec, R.; Bodnar, L.; Szczylik, C. Prognostic significance of pancreatic metastases from renal cell carcinoma in patients treated with tyrosine kinase inhibitors. Anticancer Res. 2018, 38, 359–365.
- Dudani, S.; de Velasco, G.; Wells, J.C.; Gan, C.L.; Donskov, F.; Porta, C.; Pasini, F.; Lee, J.L.; Hansen, A.; Bjarnason, G.A.; et al. Evaluation of clear cell, papillary, and chromophobe renal cell carcinoma metastasis sites and association with survival. JAMA Netw. Open 2021, 4, e22021869.
- Shin, T.J.; Song, C.; Jeong, C.W.; Kwak, C.; Seo, S.; Kang, M.; Chung, J.; Hong, S.H.; Hwang, E.C.; Park, J.Y.; et al. Metastatic renal cell carcinoma to the pancreas: Clinical features and treatment outcome. J. Surg. Oncol. 2021, 123, 204–213.
- Carril-Ajuria, L.; Santos, M.; Roldán-Romero, J.M.; Rodriguez-Antona, C.; de Velasco, G. Prognostic and predictive value of PBRM1 in clear cell renal cell carcinoma. Cancers 2019, 12, 16.
- Vilar Tabanera, A.; Munoz Munoz, P.; Molina Villar, J.M.; Gajate, P.; Sanjuanbenito, A. Surgery of pancreatic metastasis from renal cell carcinoma. Cir. Esp. 2022, 100, 50–57.
- Dobrzańska, J.; Potocki, P.; Wysocki, P.J. Do solitary pancreatic metastases of renal-cell carcinoma indicate an indolent disease with a strong indication for aggressive local treatment? A case report with literature review. Oncol. Clin. Pract. 2023. in print.
- Law, C.H.; Wei, A.C.; Hanna, S.S.; Al-Zahrani, M.; Taylor, B.R.; Greig, B.; Langer, B.; Gallinger, S. Pancreatic resection for metastatic renal cell carcinoma: Presentation, treatment and outcome. Ann. Surg. Oncol. 2003, 10, 922–926.
- Yazbek, T.; Gayet, B. The place of enucleation and enucleo-resection in the treatment of pancreatic metastasis of renal cell carcinoma. JOP 2012, 13, 433–438.
- Yoshikawa, Y.; Murakami, M.; Shimizu, J.; Yasuyama, A.; Watase, C.; Kubota, M.; Miyake, Y.; Matsuura, Y.; Kim, H.M.; Hirota, M.; et al. A case of partial pancreatectomy for recurrent metastatic renal cell carcinoma in the remnant pancreas after subtotal stomach-preserving pancreaticoduodenectomy. Gan Kagaku Ryoho 2013, 40, 1900–1902.
- Macrì, A.; Fleres, F.; Putortì, A.; Lentini, M.; Ascenti, G.; Mastrojeni, C. Relapsed metachronous pancreatic metastasis from renal cell carcinoma (RCC): Report of a case and review of literature. Ann. Ital. Chir. 2014, 85, S2239253X1402283X.
- Takeshi, A.; Mitsuhiro, I.; Hiromitsu, A.; Naoyuki, Y.; Taiichiro, S.; Hiroki, S.; Takeaki, K.; Tatsuya, S.; Futoshi, O.; Hirohiro, S.; et al. Middle-segment preserving pancreatectomy for recurrent metastasis of renal cell carcinoma after pancreatoduodenenctomy: A case report. Case Rep. Surg. 2014, 2014, 648678.
- Nihei, K.; Sakamoto, K.; Suzuki, S.; Mishina, T.; Otaki, M. A case of pancreatic metastasis of renal cell carcinoma. Gan Kagaku Ryoho 2016, 43, 2274–2276.
- Schammel, J.; Schammel, C.; Schammel, D.; Trocha, S. Renal cell carcinoma metastasis to pancreas: The aggressive nature of synchronous presentation-Case report and comprehensive review of the literature. SN Compr. Clin. Med. 2020, 2, 1272–1281.
- Itamoto, S.; Abe, T.; Oshita, A.; Hanada, K.; Nakahara, M.; Noriyuki, T. Repeat pancreatic resection for metachronous pancreatic metastasis from renal cell carcinoma: A case report. Int. J. Surg. Case Rep. 2022, 94, 107022.
- Sellner, F.; Tykalsky, N.; De Santis, M.; Pont, J.; Klimpfinger, M. Solitary and multiple isolated metastases of clear cell renal carcinoma: An indication for pancreatic surgery. Ann. Surg. Oncol. 2006, 13, 75–85.